Found a total of 10000 related content

Generating 394,760 protein representations, Harvard team develops AI model to fully understand protein context
Article Introduction:Editor | Radish Skin Understanding protein function and developing molecular therapies requires identifying the cell types in which the protein acts and dissecting the interactions between proteins. However, modeling protein interactions across biological contexts remains challenging for existing algorithms. In the latest study, researchers at Harvard Medical School developed PINNACLE, a geometric deep learning method for generating context-aware protein representations. PINNACLE leverages multi-organ single cell atlases to learn on contextualized protein interaction networks, generating 394,760 protein representations from 156 cell type contexts in 24 tissues. The study is based on the concept of “ContextualAImodelsforsingl
2024-07-26
comment 0
1142

AI designs protein 'switches' from scratch, an amazing breakthrough in protein design, David Baker's research is published in Nature
Article Introduction:Editor | KX In life, it's easy to turn on a lamp or adjust the lighting. But systems that achieve similar control of biomolecular functions are complex and poorly understood. In biology, protein functions are turned on and off in complex ways, and allosteric regulation is one of the important biological regulatory mechanisms that is essential for healthy metabolism and cell signaling. But creating allostery in synthetic protein systems has always presented significant challenges. Recently, David Baker's team at the University of Washington designed a protein that can switch between assembly and disassembly reliably and accurately through allosteric control. The researchers used AI to design new proteins that do not exist in nature and designed a variety of dynamic protein arrangements. David Baker said:
2024-08-20
comment 0
677
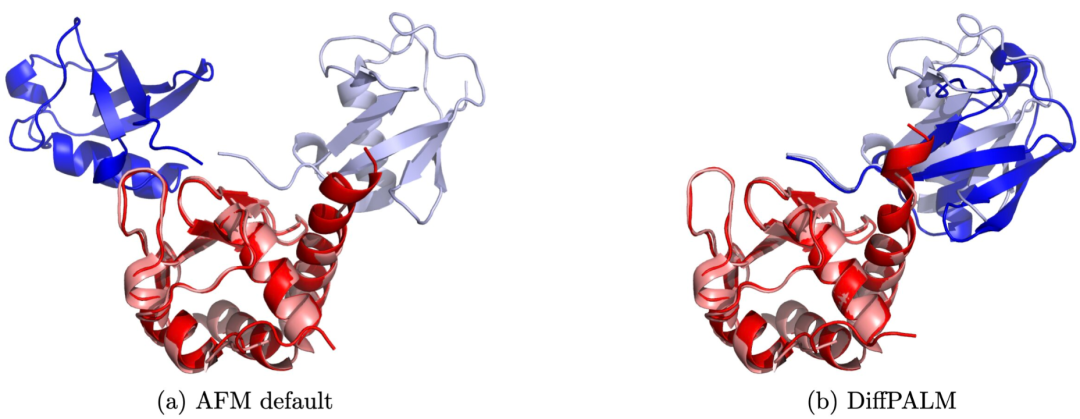
With accuracy comparable to AlphaFold, EPFL's AI method matches protein interactions from sequences
Article Introduction:1. Importance of protein interactions Proteins are the building blocks of life and participate in almost all biological processes. Understanding how proteins interact is critical to explaining the complexity of cellular function. 2. New method: pairing interacting protein sequences Anne-Florence Bitbol’s team at Ecole Polytechnique Fédérale de Lausanne (EPFL) proposed a method to pair interacting protein sequences. This method exploits the power of protein language models trained on multiple sequence alignments. 3. Method advantages This method performs well for small data sets and can improve the structure prediction of protein complexes through supervised methods. 4. The research results are published as "Pairinginteractingprotein
2024-07-16
comment 0
844
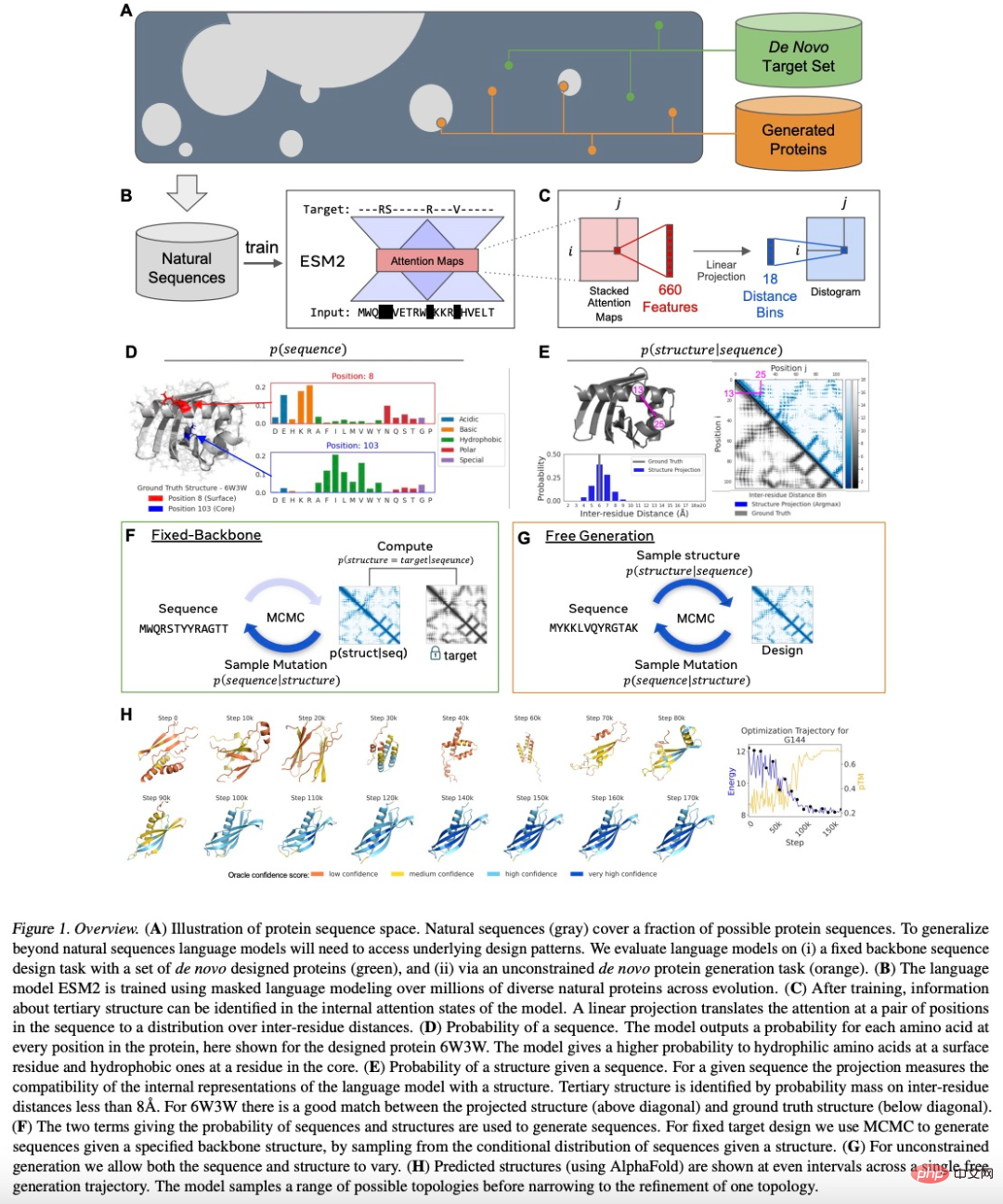

Nature sub-journal, 10 times faster, reverse protein sequence design method based on Transformer
Article Introduction:Editor | Radish Skin Protein design and engineering are advancing at an unprecedented pace thanks to advances in deep learning. However, current models cannot naturally account for non-protein entities during the design process. Here, researchers at the Ecole Polytechnique Fédérale de Lausanne (EPFL) in Switzerland propose a deep learning method based entirely on a geometric transformer of atomic coordinates and element names that can predict proteins based on backbone scaffolds with constraints imposed by different molecular environments. sequence. Using this method, researchers can produce highly thermostable, catalytically active enzymes with a high success rate. This is expected to increase the versatility of protein design pipelines to achieve desired functions. This study uses "Context-awaregeometricde
2024-08-05
comment 0
943

Meta lets a 15 billion parameter language model learn to design 'new' proteins from scratch! LeCun: Amazing results
Article Introduction:AI has once again made new progress in the biomedical field. Yes, this time it’s about protein. The difference is that in the past, AI discovered protein structures, but this time it began to design and generate protein structures on its own. If he was a "Prosecutor" in the past, it is not impossible to say that he has evolved into a "Creator" now. Participating in this research is the protein research team of FAIR, which is included in Meta's AI research institute. As the chief AI scientist who has worked at Facebook for many years, Yann LeCun also immediately forwarded the results of his team and spoke highly of it. These two papers on BioRxiv are Meta’s “amazing” achievements in protein design/generation. The system uses a simulated annealing algorithm to find an ammonia
2023-04-13
comment 0
1511

The success rate exceeds that of the RoseTTAFold series, using sequence information to directly predict protein-ligand complex structures.
Article Introduction:Editor | Radish skin protein is a well-established tool in the body's fight against pathogens and is being used to narrow down potential treatments for experimental testing. High-quality protein structure is required, and proteins are often viewed as fully or partially rigid. Here, researchers at Freie Universität Berlin have developed an artificial intelligence system that can predict fully flexible all-atom structures of protein-ligand complexes directly from sequence information. Although classical docking methods are still superior, this also depends on the crystal structure of the target protein. In addition to predicting flexible all-atom structures, the prediction confidence metric (plDDT) can be used to select accurate predictions and differentiate between strong and weak binders. The study is based on
2024-06-19
comment 0
1082

A team from the Institute of Computing Technology of the Chinese Academy of Sciences proposed CarbonNovo, an AI-based end-to-end de novo design of protein structures and sequences.
Article Introduction:Editor | ScienceAI author | Zhang Haicang team of the Institute of Computing Technology Recently, a research team led by Zhang Haicang of the Institute of Computing Technology of the Chinese Academy of Sciences proposed CarbonNovo to jointly design protein backbone structures and sequences in an end-to-end manner. The research was published at the machine learning conference ICML2024 under the title "CarbonNovo:JointDesignofProteinStructureandSequenceUsingaUnifiedEnergy-basedModel". Background Introduction Proteins are important macromolecules that perform biological functions. De novo protein design aims to create entirely new proteins and has broad applications in drug development and enzyme engineering. In recent years, basic
2024-08-21
comment 0
750

AlphaFold 3 is launched, comprehensively predicting the interactions and structures of proteins and all living molecules, with far greater accuracy than ever before
Article Introduction:Editor | Radish Skin Since the release of the powerful AlphaFold2 in 2021, scientists have been using protein structure prediction models to map various protein structures within cells, discover drugs, and draw a "cosmic map" of every known protein interaction. . Just now, Google DeepMind released the AlphaFold3 model, which can perform joint structure predictions for complexes including proteins, nucleic acids, small molecules, ions and modified residues. The accuracy of AlphaFold3 has been significantly improved compared to many dedicated tools in the past (protein-ligand interaction, protein-nucleic acid interaction, antibody-antigen prediction). This shows that within a single unified deep learning framework, it is possible to achieve
2024-07-16
comment 0
487

New SOTA for protein function prediction, statistics-based AI methods from Shanghai Institute of Technology, Oxford and others, published in Nature sub-journal
Article Introduction:, Editor | KX proteins combine with other molecules to facilitate nearly all fundamental biological activities. Therefore, understanding protein function is critical to understanding health, disease, evolution, and organismal function at the molecular level. However, more than 200 million proteins remain uncharacterized, and computational methods rely heavily on structural information of proteins to predict annotations of varying quality. Recently, a research team from the University of Oxford, ETH Zurich, the University of Shanghai for Science and Technology, and Beijing Normal University designed a statistics-based graph network method called PhiGnet to facilitate the functional annotation of proteins and the identification of functional sites. . PhiGnet not only outperforms other methods in performance, but also shrinks sequences even in the absence of structural information
2024-08-22
comment 0
745

To predict protein-DNA binding specificity, USC team develops new geometric deep learning method
Article Introduction:Editor | Radish Skin Predicting protein-DNA binding specificity is a challenging but crucial task for understanding gene regulation. Protein-DNA complexes typically bind to selected DNA targets, whereas proteins bind to a broad range of DNA sequences with varying degrees of binding specificity. This information is not directly accessible in a single structure. To access this information, researchers at the University of Southern California and the University of Washington proposed the Deep Binding Specificity Predictor (DeepPBS), a geometric deep learning model designed to predict binding based on protein-DNA structures.
2024-08-19
comment 0
1195
Simulating 500 million years of evolutionary information, it is the first large-scale biological model to simultaneously infer protein sequence, structure and function.
Article Introduction:Editor |During the **long** natural evolution process of three billion years, the **form** of the **existing** proteins was formed and went through a long natural selection process. Evolution is like a parallel experiment conducted on geological time scales, through random mutation and selection mechanisms, sifting according to the sequence, structure and function of proteins. , here researchers at EvolutionaryScale show that language models trained on evolution-generated markers can serve as evolutionary simulators for generating functional proteins that differ from known protein sequences. protein. , researchers propose the **cutting-edge** ESM3, an **advanced** multimodal generative language model that can reason about proteins
2024-06-26
comment 0
917
SOTA performance, Xiamen multi-modal protein-ligand affinity prediction AI method, combines molecular surface information for the first time
Article Introduction:Editor | KX In the field of drug research and development, accurately and effectively predicting the binding affinity of proteins and ligands is crucial for drug screening and optimization. However, current studies do not take into account the important role of molecular surface information in protein-ligand interactions. Based on this, researchers from Xiamen University proposed a novel multi-modal feature extraction (MFE) framework, which for the first time combines information on protein surface, 3D structure and sequence, and uses a cross-attention mechanism to compare different modalities. feature alignment. Experimental results demonstrate that this method achieves state-of-the-art performance in predicting protein-ligand binding affinities. Furthermore, ablation studies demonstrate the effectiveness and necessity of protein surface information and multimodal feature alignment within this framework. Related research begins with "S
2024-07-17
comment 0
1062

30 times more efficient than traditional methods, the Transformer deep learning model of the Chinese Academy of Sciences team predicts sugar-protein interaction sites
Article Introduction:Sugars are the most abundant organic substances in nature and are essential for life. Understanding how carbohydrates regulate proteins during physiological and pathological processes can provide opportunities to address key biological questions and develop new treatments. However, the diversity and complexity of sugar molecules poses a challenge to experimentally identify sugar-protein binding and interaction sites. Here, a team from the Chinese Academy of Sciences developed DeepGlycanSite, a deep learning model that can accurately predict sugar-binding sites on a given protein structure. DeepGlycanSite integrates the geometric and evolutionary characteristics of proteins into a deep equivariant graph neural network with a Transformer architecture. Its performance significantly surpasses previous advanced methods and can effectively predict
2024-07-01
comment 0
942

30 times higher than traditional methods, the Transformer deep learning model of the Chinese Academy of Sciences team predicts sugar-protein interaction sites
Article Introduction:Editor | Radish skin sugars are the most abundant organic substances in nature and are vital to life. Understanding how carbohydrates regulate proteins during physiological and pathological processes can provide opportunities to address key biological questions and develop new treatments. However, the diversity and complexity of sugar molecules poses a challenge to experimentally identify sugar-protein binding and interaction sites. Here, a team from the Chinese Academy of Sciences developed DeepGlycanSite, a deep learning model that can accurately predict sugar-binding sites on a given protein structure. DeepGlycanSite incorporates protein geometric and evolutionary features into a deep equivariant graph neural network with a Transformer architecture, significantly outperforming previous state-of-the-art methods.
2024-06-26
comment 0
1074

'AI+physics prior knowledge', Zhejiang University and Chinese Academy of Sciences general protein-ligand interaction scoring method published in Nature sub-journal
Article Introduction:Editor | X protein is like a precision lock in the body, and the drug molecule is the key. Only the key that fits perfectly can unlock the door to treatment. Scientists have been looking for efficient ways to predict the fit between these "keys" and "locks," or protein-ligand interactions. However, traditional data-driven methods often fall into "rote learning", memorizing ligand and protein training data instead of truly learning the interactions between them. Recently, a research team from Zhejiang University and the Chinese Academy of Sciences proposed a new scoring method called EquiScore, which uses heterogeneous graph neural networks to integrate physical prior knowledge and characterize protein-ligand interactions in the equation transformation space. EquiScore is trained on a new data set
2024-06-14
comment 0
1000

New method of glycoproteomics, Fudan developed a hybrid end-to-end framework based on Transformer and GNN, published in Nature sub-journal
Article Introduction:Editor | Radish skin protein glycosylation is a post-translational modification of proteins by sugar groups, which plays an important role in various physiological and pathological functions of cells. Glycoproteomics is the study of protein glycosylation within the proteome, using liquid chromatography coupled with tandem mass spectrometry (MS/MS) technology to obtain combined information on glycosylation sites, glycosylation levels and sugar structures. However, current database search methods for glycoproteomics often struggle to determine glycan structures due to the limited occurrence of structure-determining ions. Although spectral search methods can exploit fragmentation intensity to facilitate structural identification of glycopeptides, difficulties in spectral library construction hinder their application. In the latest research, researchers from Fudan University proposed DeepGP, a Transform-based
2024-08-06
comment 0
426

Guide to the location of white-fronted wild goose eggs in 'The Condor Shooting'
Article Introduction:The White-fronted Goose Egg is an important synthetic material in the Condor Mobile Game. Many players want to know where the White-fronted Goose Egg is in the Condor Mobile Game. The White-fronted Goose Egg is in the second and third places of Wudingfang Lingshuiyuan in Zhongdu. On the eaves of a house, the editor will introduce to you in detail the location strategy of the white-fronted wild goose egg in "The Condor Shooting". Players who are interested must not miss it! The strategy for the location of the white-fronted wild goose egg in "The Condor Shooting": 1. First, go to [Wudingfang Lingshuiyuan in Zhongdu] and jump on the first roof; 2. Continue to jump on the second eaves and follow the light spot to find the first [White-fronted Wild Goose Egg]; 3. Continue going up. Jump, jump up to the third eaves, follow the light spots to find the second and third [White-fronted Goose Eggs]; 4. White-fronted Goose Eggs are an important prop for synthesizing the realm material [Sparrow Saliva].
2024-03-31
comment 0
1398

