Insgesamt10000 bezogener Inhalt gefunden

Das Harvard-Team generiert 394.760 Proteindarstellungen und entwickelt ein KI-Modell, um den Proteinkontext vollständig zu verstehen
Artikeleinführung:Herausgeber | Rettichhaut Um die Funktion von Proteinen zu verstehen und molekulare Therapien zu entwickeln, müssen die Zelltypen identifiziert werden, in denen das Protein wirkt, und die Wechselwirkungen zwischen Proteinen analysiert werden. Allerdings bleibt die Modellierung von Proteininteraktionen über biologische Kontexte hinweg eine Herausforderung für bestehende Algorithmen. In der neuesten Studie entwickelten Forscher der Harvard Medical School PINNACLE, eine geometrische Deep-Learning-Methode zur Generierung kontextbezogener Proteindarstellungen. PINNACLE nutzt Multiorgan-Einzelzellatlanten, um kontextualisierte Proteininteraktionsnetzwerke zu erlernen und generiert 394.760 Proteindarstellungen aus 156 Zelltypkontexten in 24 Geweben. Die Studie basiert auf dem Konzept „ContextualAImodelsforsingl“.
2024-07-26
Kommentar 0
1142

KI entwirft Protein-„Schalter' von Grund auf, ein erstaunlicher Durchbruch im Proteindesign. David Bakers Forschung wird in Nature veröffentlicht
Artikeleinführung:Editor |. KX Im Leben ist es einfach, eine Lampe einzuschalten oder die Beleuchtung anzupassen. Doch Systeme, die eine ähnliche Kontrolle biomolekularer Funktionen erreichen, sind komplex und wenig verstanden. In der Biologie werden Proteinfunktionen auf komplexe Weise ein- und ausgeschaltet, und die allosterische Regulierung ist einer der wichtigen biologischen Regulierungsmechanismen, die für einen gesunden Stoffwechsel und die Zellsignalisierung unerlässlich sind. Aber die Herstellung von Allosterie in synthetischen Proteinsystemen war schon immer mit großen Herausforderungen verbunden. Kürzlich hat das Team von David Baker an der University of Washington ein Protein entwickelt, das durch allosterische Kontrolle zuverlässig und genau zwischen Auf- und Abbau wechseln kann. Die Forscher nutzten KI, um neue Proteine zu entwerfen, die in der Natur nicht vorkommen, und entwarfen eine Vielzahl dynamischer Proteinanordnungen. David Baker sagte:
2024-08-20
Kommentar 0
677
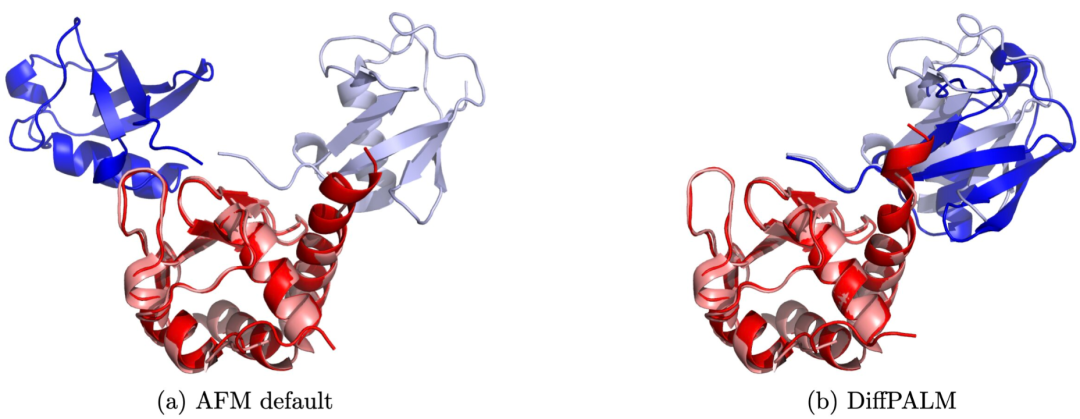
Mit einer mit AlphaFold vergleichbaren Genauigkeit gleicht die KI-Methode der EPFL Proteininteraktionen aus Sequenzen ab
Artikeleinführung:1. Bedeutung von Proteininteraktionen Proteine sind die Bausteine des Lebens und an nahezu allen biologischen Prozessen beteiligt. Um die Komplexität der Zellfunktion zu erklären, ist es wichtig zu verstehen, wie Proteine interagieren. 2. Neue Methode: Paarung interagierender Proteinsequenzen Anne-Florence Bitbols Team an der Ecole Polytechnique Fédérale de Lausanne (EPFL) schlug eine Methode zur Paarung interagierender Proteinsequenzen vor. Diese Methode nutzt die Leistungsfähigkeit von Proteinsprachmodellen, die auf mehreren Sequenzausrichtungen trainiert wurden. 3. Vorteile der Methode Diese Methode eignet sich gut für kleine Datensätze und kann die Strukturvorhersage von Proteinkomplexen durch überwachte Methoden verbessern. 4. Die Forschungsergebnisse werden unter dem Titel „Pairinginteractingprotein“ veröffentlicht
2024-07-16
Kommentar 0
844

Nature-Unterjournal, 10-mal schnellere, umgekehrte Proteinsequenz-Designmethode basierend auf Transformer
Artikeleinführung:Herausgeber |. Das Design und die Entwicklung von Radish Skin Protein schreiten dank der Fortschritte im Deep Learning in beispiellosem Tempo voran. Aktuelle Modelle können jedoch nicht auf natürliche Weise Nicht-Protein-Entitäten während des Designprozesses berücksichtigen. Hier schlagen Forscher der Ecole Polytechnique Fédérale de Lausanne (EPFL) in der Schweiz eine Deep-Learning-Methode vor, die vollständig auf einem geometrischen Transformator von Atomkoordinaten und Elementnamen basiert und Proteine basierend auf Grundgerüsten mit Einschränkungen durch unterschiedliche molekulare Umgebungen vorhersagen kann. Mit dieser Methode können Forscher hoch thermostabile, katalytisch aktive Enzyme mit hoher Erfolgsquote herstellen. Es wird erwartet, dass dies die Vielseitigkeit von Proteindesign-Pipelines zur Erreichung gewünschter Funktionen erhöht. Diese Studie verwendet „Context-awaregeometricde“.
2024-08-05
Kommentar 0
943

Meta lässt ein Sprachmodell mit 15 Milliarden Parametern lernen, „neue' Proteine von Grund auf zu entwerfen! LeCun: Erstaunliche Ergebnisse
Artikeleinführung:Die KI hat im biomedizinischen Bereich erneut neue Fortschritte erzielt. Ja, dieses Mal geht es um Protein. Der Unterschied besteht darin, dass die KI in der Vergangenheit Proteinstrukturen entdeckte, dieses Mal jedoch begann, Proteinstrukturen selbst zu entwerfen und zu erzeugen. Wenn er in der Vergangenheit ein „Staatsanwalt“ war, ist es nicht unmöglich zu sagen, dass er sich jetzt zu einem „Schöpfer“ entwickelt hat. An dieser Forschung ist das Proteinforschungsteam von FAIR beteiligt, das zum KI-Forschungsinstitut von Meta gehört. Als leitender KI-Wissenschaftler, der viele Jahre bei Facebook gearbeitet hat, leitete Yann LeCun auch sofort die Ergebnisse seines eigenen Teams weiter und lobte sie. Diese beiden Artikel über BioRxiv sind Metas „erstaunliche“ Errungenschaften im Bereich Proteindesign/-generierung. Das System verwendet einen simulierten Glühalgorithmus, um Ammoniak zu finden
2023-04-13
Kommentar 0
1511

Die Erfolgsquote übertrifft die der RoseTTAFold-Reihe, da sie Sequenzinformationen nutzt, um die Struktur von Protein-Ligand-Komplexen direkt vorherzusagen.
Artikeleinführung:Herausgeber | Rettich-Hautprotein ist ein bewährtes Mittel im Kampf des Körpers gegen Krankheitserreger und wird zur Eingrenzung potenzieller Behandlungsmethoden für experimentelle Tests eingesetzt. Es ist eine hochwertige Proteinstruktur erforderlich, und Proteine werden oft als vollständig oder teilweise starr angesehen. Hier haben Forscher der Freien Universität Berlin ein künstliches Intelligenzsystem entwickelt, das vollständig flexible Gesamtatomstrukturen von Protein-Ligand-Komplexen direkt aus Sequenzinformationen vorhersagen kann. Obwohl klassische Docking-Methoden immer noch überlegen sind, hängt dies auch von der Kristallstruktur des Zielproteins ab. Zusätzlich zur Vorhersage flexibler Allatomstrukturen kann die Vorhersagekonfidenzmetrik (plDDT) verwendet werden, um genaue Vorhersagen auszuwählen und zwischen starken und schwachen Bindemitteln zu unterscheiden. Die Studie basiert auf
2024-06-19
Kommentar 0
1082


AlphaFold 3 wird auf den Markt gebracht und sagt die Wechselwirkungen und Strukturen von Proteinen und allen Lebensmolekülen umfassend und mit weitaus größerer Genauigkeit als je zuvor voraus
Artikeleinführung:Herausgeber | Rettichhaut Seit der Veröffentlichung des leistungsstarken AlphaFold2 im Jahr 2021 verwenden Wissenschaftler Modelle zur Proteinstrukturvorhersage, um verschiedene Proteinstrukturen innerhalb von Zellen zu kartieren, Medikamente zu entdecken und eine „kosmische Karte“ jeder bekannten Proteininteraktion zu zeichnen. Gerade hat Google DeepMind das AlphaFold3-Modell veröffentlicht, das gemeinsame Strukturvorhersagen für Komplexe wie Proteine, Nukleinsäuren, kleine Moleküle, Ionen und modifizierte Reste durchführen kann. Die Genauigkeit von AlphaFold3 wurde im Vergleich zu vielen dedizierten Tools in der Vergangenheit (Protein-Ligand-Interaktion, Protein-Nukleinsäure-Interaktion, Antikörper-Antigen-Vorhersage) deutlich verbessert. Dies zeigt, dass dies innerhalb eines einzigen einheitlichen Deep-Learning-Frameworks möglich ist
2024-07-16
Kommentar 0
487

Neues SOTA für die Vorhersage von Proteinfunktionen, statistikbasierte KI-Methoden vom Shanghai Institute of Technology, Oxford und anderen, veröffentlicht im Nature-Unterjournal
Artikeleinführung:, Herausgeber |. KX-Proteine verbinden sich mit anderen Molekülen, um nahezu alle grundlegenden biologischen Aktivitäten zu ermöglichen. Daher ist das Verständnis der Proteinfunktion von entscheidender Bedeutung für das Verständnis von Gesundheit, Krankheit, Evolution und Organismusfunktion auf molekularer Ebene. Allerdings sind mehr als 200 Millionen Proteine noch nicht charakterisiert, und Berechnungsmethoden stützen sich in hohem Maße auf Strukturinformationen von Proteinen, um Annotationen unterschiedlicher Qualität vorherzusagen. Kürzlich hat ein Forschungsteam der Universität Oxford, der ETH Zürich, der Universität Shanghai für Wissenschaft und Technologie und der Beijing Normal University eine statistikbasierte Graphnetzwerkmethode namens PhiGnet entwickelt, um die funktionelle Annotation von Proteinen und die Identifizierung funktioneller Stellen zu erleichtern . . PhiGnet übertrifft nicht nur andere Methoden in der Leistung, sondern verkleinert auch Sequenzen, selbst wenn keine Strukturinformationen vorliegen
2024-08-22
Kommentar 0
745

Um die Protein-DNA-Bindungsspezifität vorherzusagen, entwickelt das USC-Team eine neue geometrische Deep-Learning-Methode
Artikeleinführung:Herausgeber | Rettichhaut Die Vorhersage der Protein-DNA-Bindungsspezifität ist eine anspruchsvolle, aber entscheidende Aufgabe für das Verständnis der Genregulation. Protein-DNA-Komplexe binden typischerweise an ausgewählte DNA-Ziele, wohingegen Proteine an ein breites Spektrum von DNA-Sequenzen mit unterschiedlichem Grad an Bindungsspezifität binden. Diese Informationen sind nicht direkt in einer einzelnen Struktur zugänglich. Um auf diese Informationen zuzugreifen, schlugen Forscher der University of Southern California und der University of Washington den Deep Binding Specificity Predictor (DeepPBS) vor, ein geometrisches Deep-Learning-Modell, das die Bindung anhand von Protein-DNA-Strukturen vorhersagen soll.
2024-08-19
Kommentar 0
1195
Es simuliert evolutionäre Informationen aus 500 Millionen Jahren und ist das erste groß angelegte biologische Modell, das gleichzeitig auf Proteinsequenz, -struktur und -funktion schließen kann.
Artikeleinführung:Herausgeber |Während der **langen** drei Milliarden Jahre der natürlichen Evolution wurde die **Form** der **existierenden** Proteine gebildet und durchlief einen langen natürlichen Selektionsprozess. Evolution ist wie ein Parallelexperiment, das auf geologischen Zeitskalen durch zufällige Mutations- und Selektionsmechanismen durchgeführt wird und nach der Reihenfolge, Struktur und Funktion von Proteinen sortiert wird. Hier zeigen Forscher von EvolutionaryScale, dass Sprachmodelle, die auf evolutionär erzeugten Markern trainiert wurden, als Evolutionssimulatoren für die Erzeugung funktioneller Proteine dienen können, die sich von bekannten Proteinsequenzen unterscheiden. Forscher schlagen das **modernste** ESM3 vor, ein **fortgeschrittenes** multimodales generatives Sprachmodell, das über Proteine schlussfolgern kann
2024-06-26
Kommentar 0
917
SOTA Performance, eine multimodale KI-Methode zur Vorhersage der Protein-Ligand-Affinität in Xiamen, kombiniert erstmals molekulare Oberflächeninformationen
Artikeleinführung:Herausgeber |. KX Im Bereich der Arzneimittelforschung und -entwicklung ist die genaue und effektive Vorhersage der Bindungsaffinität von Proteinen und Liganden für das Arzneimittelscreening und die Arzneimitteloptimierung von entscheidender Bedeutung. Aktuelle Studien berücksichtigen jedoch nicht die wichtige Rolle molekularer Oberflächeninformationen bei Protein-Ligand-Wechselwirkungen. Auf dieser Grundlage schlugen Forscher der Universität Xiamen ein neuartiges Framework zur multimodalen Merkmalsextraktion (MFE) vor, das erstmals Informationen über Proteinoberfläche, 3D-Struktur und -Sequenz kombiniert und einen Kreuzaufmerksamkeitsmechanismus verwendet, um verschiedene Modalitäten zu vergleichen Ausrichtung. Experimentelle Ergebnisse zeigen, dass diese Methode bei der Vorhersage von Protein-Ligand-Bindungsaffinitäten Spitzenleistungen erbringt. Darüber hinaus belegen Ablationsstudien die Wirksamkeit und Notwendigkeit der Proteinoberflächeninformation und der multimodalen Merkmalsausrichtung innerhalb dieses Rahmens. Verwandte Forschungen beginnen mit „S
2024-07-17
Kommentar 0
1062

Das Transformer-Deep-Learning-Modell des Teams der Chinesischen Akademie der Wissenschaften ist 30-mal effizienter als herkömmliche Methoden und sagt die Interaktionsorte zwischen Zucker und Protein voraus
Artikeleinführung:Zucker ist die am häufigsten vorkommende organische Substanz in der Natur und lebensnotwendig. Das Verständnis, wie Kohlenhydrate Proteine während physiologischer und pathologischer Prozesse regulieren, kann Möglichkeiten bieten, wichtige biologische Fragen zu beantworten und neue Behandlungen zu entwickeln. Die Vielfalt und Komplexität von Zuckermolekülen stellt jedoch eine Herausforderung dar, wenn es darum geht, Bindungs- und Interaktionsstellen zwischen Zucker und Protein experimentell zu identifizieren. Hier entwickelte ein Team der Chinesischen Akademie der Wissenschaften DeepGlycanSite, ein Deep-Learning-Modell, das Zuckerbindungsstellen auf einer bestimmten Proteinstruktur genau vorhersagen kann. DeepGlycanSite integriert die geometrischen und evolutionären Eigenschaften von Proteinen in ein tiefes äquivariantes graphisches neuronales Netzwerk mit einer Transformer-Architektur. Seine Leistung übertrifft frühere fortschrittliche Methoden deutlich und kann effektiv vorhersagen
2024-07-01
Kommentar 0
942

Das Transformer-Deep-Learning-Modell des Teams der Chinesischen Akademie der Wissenschaften ist 30-mal besser als herkömmliche Methoden und sagt die Interaktionsorte zwischen Zucker und Protein voraus
Artikeleinführung:Herausgeber |. Rettichschalenzucker ist der am häufigsten vorkommende organische Stoff in der Natur und lebenswichtig. Das Verständnis, wie Kohlenhydrate Proteine während physiologischer und pathologischer Prozesse regulieren, kann Möglichkeiten bieten, wichtige biologische Fragen zu beantworten und neue Behandlungen zu entwickeln. Die Vielfalt und Komplexität von Zuckermolekülen stellt jedoch eine Herausforderung dar, wenn es darum geht, Bindungs- und Interaktionsstellen zwischen Zucker und Protein experimentell zu identifizieren. Hier entwickelte ein Team der Chinesischen Akademie der Wissenschaften DeepGlycanSite, ein Deep-Learning-Modell, das Zuckerbindungsstellen auf einer bestimmten Proteinstruktur genau vorhersagen kann. DeepGlycanSite integriert Proteingeometrie und evolutionäre Merkmale in ein tiefes äquivariantes graphisches neuronales Netzwerk mit einer Transformer-Architektur und übertrifft damit bisherige Methoden auf dem neuesten Stand der Technik deutlich.
2024-06-26
Kommentar 0
1074

„KI+Physik-Vorkenntnisse', allgemeine Bewertungsmethode für Protein-Ligand-Interaktionen der Zhejiang-Universität und der Chinesischen Akademie der Wissenschaften, veröffentlicht im Nature-Unterjournal
Artikeleinführung:Herausgeber |. Wissenschaftler haben nach effizienten Möglichkeiten gesucht, die Passung zwischen diesen „Schlüsseln“ und „Schlössern“ oder Protein-Ligand-Wechselwirkungen vorherzusagen. Traditionelle datengesteuerte Methoden verfallen jedoch häufig in das „Auswendiglernen“ und merken sich Liganden- und Protein-Trainingsdaten, anstatt die Wechselwirkungen zwischen ihnen wirklich zu lernen. Kürzlich hat ein Forschungsteam der Zhejiang-Universität und der Chinesischen Akademie der Wissenschaften eine neue Bewertungsmethode namens EquiScore vorgeschlagen, die heterogene graphische neuronale Netze nutzt, um physikalisches Vorwissen zu integrieren und Protein-Ligand-Wechselwirkungen im Gleichungstransformationsraum zu charakterisieren. EquiScore wird anhand eines neuen Datensatzes trainiert
2024-06-14
Kommentar 0
1000

Als neue Methode der Glykoproteomik entwickelte Fudan ein hybrides End-to-End-Framework auf Basis von Transformer und GNN, veröffentlicht im Nature-Unterjournal
Artikeleinführung:Herausgeber |. Die Glykosylierung von Rettichhautproteinen ist eine posttranslationale Modifikation von Proteinen durch Zuckergruppen, die eine wichtige Rolle bei verschiedenen physiologischen und pathologischen Funktionen von Zellen spielt. Glykoproteomik ist die Untersuchung der Proteinglykosylierung innerhalb des Proteoms, wobei Flüssigkeitschromatographie in Verbindung mit Tandem-Massenspektrometrie-Technologie (MS/MS) zum Einsatz kommt, um kombinierte Informationen über Glykosylierungsstellen, Glykosylierungsniveaus und Zuckerstrukturen zu erhalten. Aktuelle Datenbanksuchmethoden für die Glykoproteomik haben jedoch aufgrund des begrenzten Vorkommens strukturbestimmender Ionen oft Schwierigkeiten, Glykanstrukturen zu bestimmen. Obwohl spektrale Suchmethoden die Fragmentierungsintensität nutzen können, um die strukturelle Identifizierung von Glykopeptiden zu erleichtern, behindern Schwierigkeiten beim Aufbau der Spektralbibliothek ihre Anwendung. In der neuesten Forschung schlugen Forscher der Fudan-Universität DeepGP vor, ein auf Transformationen basierendes Verfahren
2024-08-06
Kommentar 0
426

Leitfaden zum Standort der Blässgänseeier in „The Condor Shooting'
Artikeleinführung:Das Weißstirn-Gänseei ist ein wichtiges synthetisches Material im Condor-Handyspiel. Viele Spieler möchten wissen, wo sich das Weißstirn-Gänseei im Condor-Handyspiel befindet Wudingfang Lingshuiyuan in Zhongdu wird Ihnen in „The Condor Shooting“ ausführlich die Standortstrategie des Weißstirn-Gänseeis vorstellen Strategie des Blässgans-Eies in „The Condor Shooting“: 1. Gehen Sie zuerst zu [Wudingfang Lingshuiyuan in Zhongdu] und springen Sie auf das erste Dach. 2. Springen Sie weiter auf die zweite Dachtraufe und folgen Sie dem Lichtpunkt Finden Sie das erste [Blässgans-Ei] 3. Gehen Sie weiter nach oben. Springen Sie zur dritten Traufe, folgen Sie den Lichtpunkten, um das zweite und dritte [Blässgans-Ei] zu finden Gänseeier sind ein wichtiges Hilfsmittel zur Synthese des Reichsmaterials [Sparrow Speichel].
2024-03-31
Kommentar 0
1398

