


Drug discovery is a complex, multi-step process involving the intersection of many subdisciplines of chemistry and biology. Human medicinal chemists play an important role in this process with their years of accumulated expertise
So, can artificial intelligence (AI) fill the role that medicinal chemists play in drug discovery? The answer may be yes.
Recently, a research team from the Novartis Institutes for Biomedical Research (NIBR) and the Microsoft Research Center for Scientific Intelligence (AI4Science) jointly proposed a machine learning model that can partially reproduce the work of professional chemists. The collective knowledge accumulated among scientists is often referred to as "chemical intuition."
The research team believes that this method can be used as a complement to molecular modeling to improve the efficiency of future drug development
The research paper is titled "Extracting Intuition in Medicinal Chemistry through Preference Machine Learning" and has been published in Nature Communications, a sub-journal of Nature

Machine learning recreates medicinal chemist expertise
Medicinal chemists, both wet lab and computational, play a crucial role in the "lead optimization" phase of drug discovery, as they are often asked to determine which compounds need to be synthesized and subsequently optimized. Evaluation takes place during rounds.
To do this, medicinal chemists typically review data including compound properties such as activity, ADMET2, or target structural information. Therefore, the success of a project depends not only on the quality of the experimental data generated, but also on the robustness and rationality of the decisions made by the team working in medicinal chemistry.
Medicinal chemists are able to make decisions more efficiently because they often draw on expertise to have an intuitive understanding of what succeeds in different iterations of early-stage drug discovery.
While there have been previous attempts to formalize this knowledge using rule-based approaches or simple cheminformatic feasibility scores, capturing the subtleties and complexities involved in scoring by medicinal chemists remains a fundamental Challenge
To achieve this goal, the research aims to transform expert knowledge into part of a machine learning model. This model can be used as an auxiliary tool, like other recommendation systems that have been reported in the industry, in the deployment of decision-making processes in lead optimization or other aspects of drug discovery
Considering that medicinal chemistry currently mainly relies on manual work, it is inevitably affected by subjective bias. Some studies have reported low agreement in ratings between medicinal chemists as well as within medicinal chemists. In this study, the researchers hope to solve some problems by borrowing strategies from multiplayer games.
They viewed the task of ranking a set of molecules as a preference learning problem and then used a simple neural network to model individual preferences
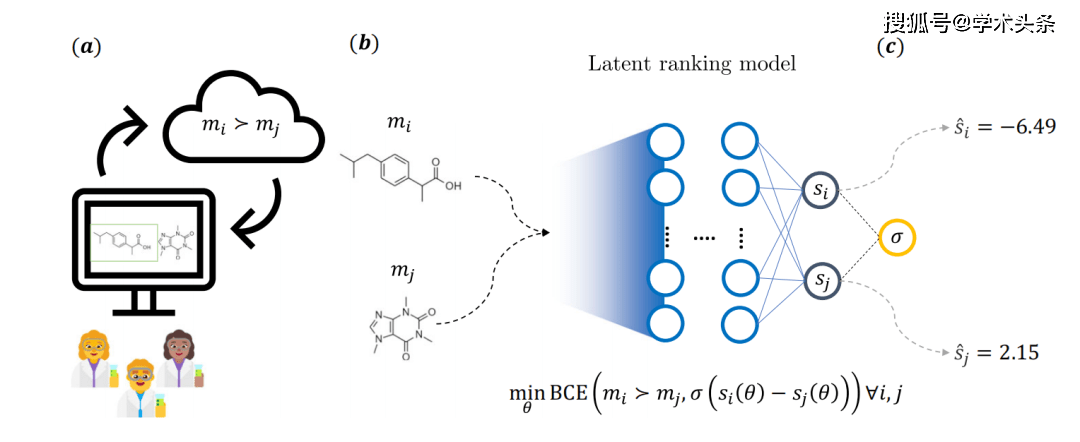 Figure | Overall schematic diagram of the main ideas of the research (source: the paper)
Figure | Overall schematic diagram of the main ideas of the research (source: the paper)
Specifically, as shown in the figure above, molecules are viewed as participants in a competitive game, with the probability of one side winning determined by feedback provided by the chemist. To do this, medicinal chemists answer pre-specified question prompts on a web application and select one of two molecules. A total of 35 Novartis medicinal chemists were involved in the process, resulting in a total of more than 5000 annotations.
This feedback led to an implicit scoring model, which uses a model with two independent neural network structures. Each branch has a fixed weight and the molecules are characterized using common chemoinformatics descriptors. During training, the model's parameters are optimized via a binary cross-entropy loss (BCE loss), which depends on the underlying score difference of a pair of molecules and feedback provided by the chemist
Once training is complete, the score for any arbitrary molecule can be inferred, which can then be used for downstream cheminformatics tasks.
In addition, the model can more accurately determine the similarities between different drugs. The learning scoring function proposed in the study is more accurate than the traditional drug similarity evaluation index (QED)
It is worth noting that In order to promote the reproducibility of the research and further development of the field, the researchers also provide a software package called "MolSkill", which contains the model and anonymization response data.
Problems and applications of machine learning in the field of medicinal chemistry
However, although this model can reproduce the knowledge accumulated by medicinal chemists in their work, it also has some limitations. First, in order to capture chemical intuition, the questions asked during data collection have always been vague.
Also, although the proposed study design resulted in greater agreement between participants compared with previous studies, the pairwise comparison method is not perfect.
In addition, the "Flatland fallacy" leads to the human tendency to simplify high-dimensional problems into a small set of variables that can be cognitively tracked, and this simplification may be affected by the personal characteristics of each medicinal chemist
However, the research team stated that the model proposed in this study is not limited to the scope of application of the current study. Specifically, the framework discussed can be extended to other quantifiable but expensive observables in the field of drug discovery. Furthermore, it can provide insights into as yet unexplored areas of chemical space.
Taking this into account, the research team believes that some popular rule-based filters can learn from artificially generated training data to build a similar architecture. This model can overcome the major limitation of having to manually filter compounds before making inferences
The same approach can also be used to generate compound scores that prioritize combinations in synthetic chemical libraries where screening due to their natural novelty is difficult using existing rule-based methods
Another thing that needs to be re-expressed is: in a prospective, primary optimization scenario for a specific target, multiple sources of information (such as biological properties, ADMET, etc.) need to be comprehensively considered to test the validity of the research framework. Practicality
The research team wrote in the paper: "Machine learning methods can design thousands of compounds, and techniques such as high-throughput screening can highlight a large number of candidate compounds in the early stages of the drug discovery process. The score proposed this time Methods are being used to implicitly incorporate chemists’ intuition to screen compounds without the need for manual inspection. The expectation is that this application will accelerate method adoption and trust in the coming years.”
The above is the detailed content of AI drug researcher joins Nature sub-journal: using professional knowledge to accelerate drug development. For more information, please follow other related articles on the PHP Chinese website!
















![[Web front-end] Node.js quick start](https://img.php.cn/upload/course/000/000/067/662b5d34ba7c0227.png)



