


In recent years, with the rapid development of single cell technology, we have been able to measure various characteristics of single cells to obtain single cell multi-modal data (such as scRNA-seq, scATAC-seq, Patch-seq) .
These data help us gain insights into cellular functions and molecular mechanisms. For example, researchers have recently used machine learning methods to analyze the relationship between single-cell multi-modal data to understand the biological mechanisms involved in cell types and diseases.
However, the acquisition of single-cell multi-modal data is often costly, and modal loss often occurs. Existing machine learning methods usually require fully matched multi-modal data for data filling and embedding, and are not suitable for situations where modalities are missing.
In order to solve this problem, Wang Daifeng's laboratory at the University of Wisconsin-Madison developed an open source machine learning method based on joint variational autoencoders - Joint Variational Autoencoders for Multimodal Imputation and Embedding (JAMIE).
JAMIE can be used for integrated analysis of single-cell multi-modal data, such as data alignment, embedding, and filling in missing data to better predict cell types and functions.
This work was recently published in Nature Machine Intelligence.

Paper address: https://www.nature.com/articles/s42256-023-00663 -z
Project address: https://github.com/daifengwanglab/JAMIE
JAMIE trains a reusable joint variational autoencoder model to enhance single-modal pattern inference by projecting available multimodal data separately into similar latent spaces ability.
As shown in Figure 1, to perform cross-modal imputation, JAMIE feeds data into an encoder and then processes the latent space results through the opposite decoder.
JAMIE combines the reusable and flexible latent space generation of autoencoders with the automatic correspondence estimation of alignment methods, enabling the processing of multi-modal data with incomplete correspondence.
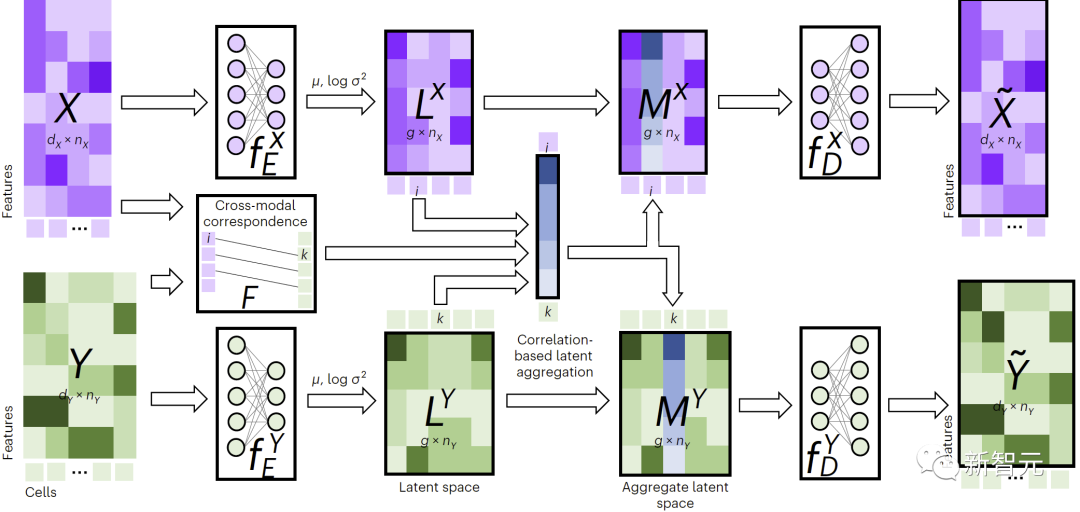
Figure 1. Overview of JAMIE method
Specifically, JAMIE can be divided into the following two steps:
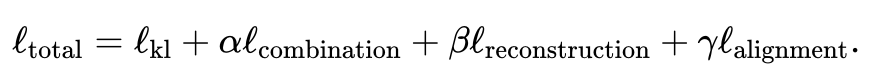
The total loss function contains four items.
The first item calculates the Kullback-Leibler (KL) divergence between the distribution inferred by the variational autoencoder and the multivariate standard normal distribution, which helps to preserve the latent space the continuity of space.
For specific expressions of each item, see the original text of the paper. The weights of the second, third, and fourth items relative to the first item can be adjusted by the user. JAMIE also provides default weights suitable for common situations.
The following table shows the comparison of the model and applicable scope of JAMIE with the current state-of-the-art methods. JAMIE unifies the features of several different integration and interpolation methods into a single architecture, thereby enabling missing modality interpolation, resulting in non-omics data compatibility and the ability to handle multimodal data with only partial correspondence. The advantages.
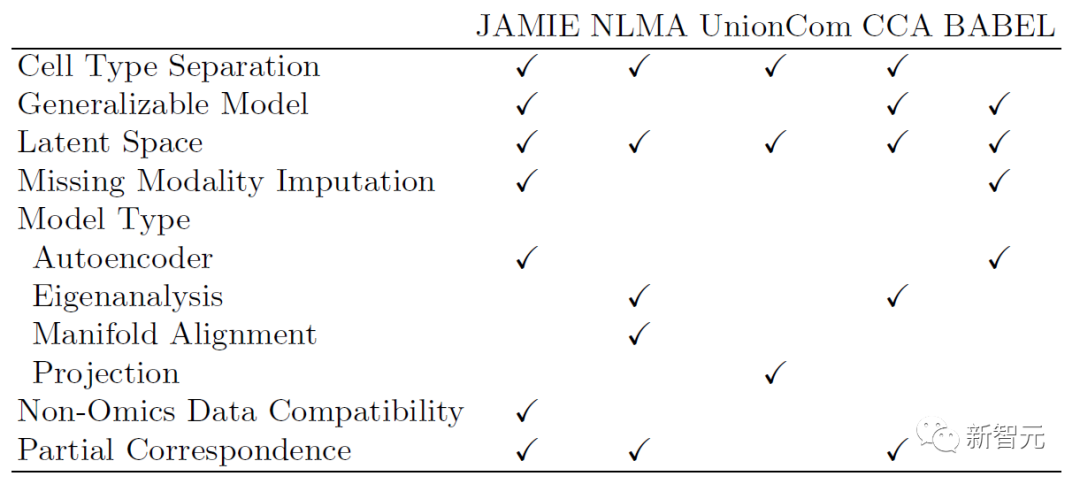
Table 1. Comparison of various multimodal integration and missing modal filling methods. Through a single architecture, JAMIE integrates features from multiple different integration and interpolation methods. NLMA: Nonlinear Manifold Alignment [15], UnionCom [7], CCA: Canonical Correlation Analysis [15, 16], BABEL [5].
Multimodal data integration and phenotype prediction
Integration of multimodal data can improve classification performance, enhance phenotypic knowledge and understanding of complex biological mechanisms.
Given two data sets , and corresponding relationships, JAMIE can generate latent space data , based on the trained encoder and , and perform clustering or classification based on .
Clustering based on latent space data has several advantages, such as incorporating both modalities into feature generation. JAMIE can then predict sample correspondences, such as cell type prediction.
For partially labeled data sets, cells in the same cluster should have similar types.
JAMIE separates the characteristics of different types of data in the process of generating latent space data, so complex clustering or classification algorithms are usually not required to achieve better results. .
For high-dimensional data, JAMIE uses UMAP [32] for cell type clustering visualization.
Cross-modal data filling
Currently many methods of cross-modal filling cannot Demonstrate that they learned underlying biological mechanisms for padding purposes.
Compared with feed-forward networks or linear regression methods, JAMIE can better learn underlying biological mechanisms to predict missing data based on a more rigorous mathematical foundation.
Figure 2 shows JAMIE’s process for cross-modal data filling. JAMIE first trains the encoding and decoding models on the training data.
For new data , JAMIE first uses the encoder learned from the data to project it into the latent space to obtain , then obtains by aggregating latent space features, and finally uses the corresponding decoder to Data decoded into missing patterns.
JAMIE uses latent space to predict the correspondence between cells, which may help understand the relationship between data features and phenotypes.
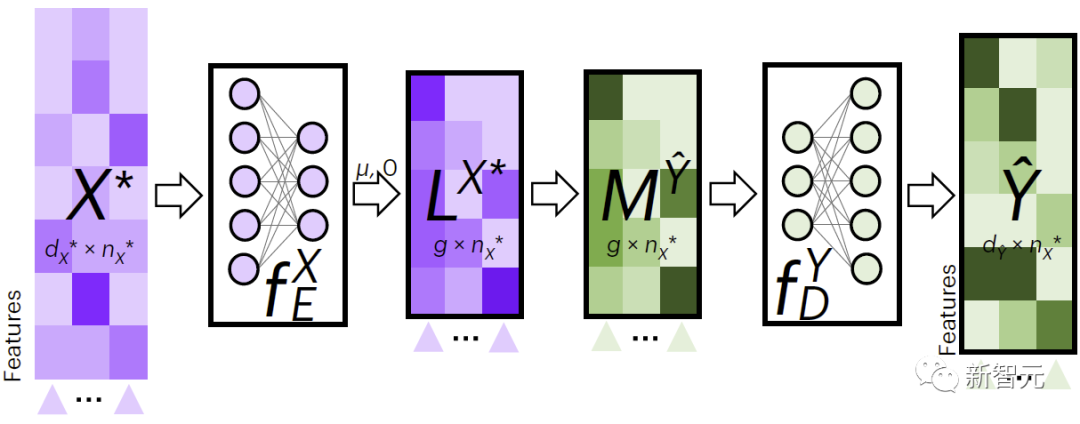
Figure 2. JAMIE cross-modal interpolation
Explanation of latent space features and filling features
In order to explain the trained model, JAMIE used SHAP (SHapley Additive exPlanations )[18].
SHAP evaluates the importance of individual input features by sample modulating the individual predictions generated by the model. This can be used for a variety of interesting applications.
If the target variable can be easily separated by phenotype, SHAP can identify relevant characteristics for further study. Furthermore, if we perform imputation, SHAP can reveal the cross-modal connections learned by the model.
Given a model and a sample, learn the SHAP value such that where is the background feature vector.
If , then the sum of the SHAP values and the background output will be equal to , where each is proportional to the impact on the model output.
Another useful technique is to select a key metric for classification (e.g., LTA [7,19]) or imputation (e.g., correspondence between imputed features and measured features relationship) and evaluate the metric by removing (replacing with background values) each feature one by one in the model.
Then, if the key metric gets worse, this indicates that the removed feature is more important to the model’s results.
JAMIE used four commonly used single-cell multi-modal data sets for verification.
(1) Simulated multimodal data (300 samples, 3 cell types) generated by Gaussian distribution sampling of branched manifolds from MMD-MA;
(2) Patch-seq genes from single neuronal cells in mouse visual cortex (3,654 samples, 6 cell types) and mouse motor cortex (1,208 samples, 9 cell types) Expression and electrophysiological signature data;
(3) 8,981 samples from the human developing brain (21 gestational weeks, 7 major cell types covering the human cerebral cortex) 10x single-cell multi-omics gene expression and chromatin accessibility data;
(4) scRNA-seq genes from 4,301 cells of the COLO-320DM colon adenocarcinoma cell line Expression and scATAC-seq chromatin accessibility data.
The evaluation found that JAMIE is significantly better than other methods (comparison of branch manifold simulation data results of MMD-MA in Figure 3, and comparison of mouse visual cortex data results in Figure 4) and Important features of multimodal filling are prioritized while providing potentially new mechanistic insights at the cellular resolution level.
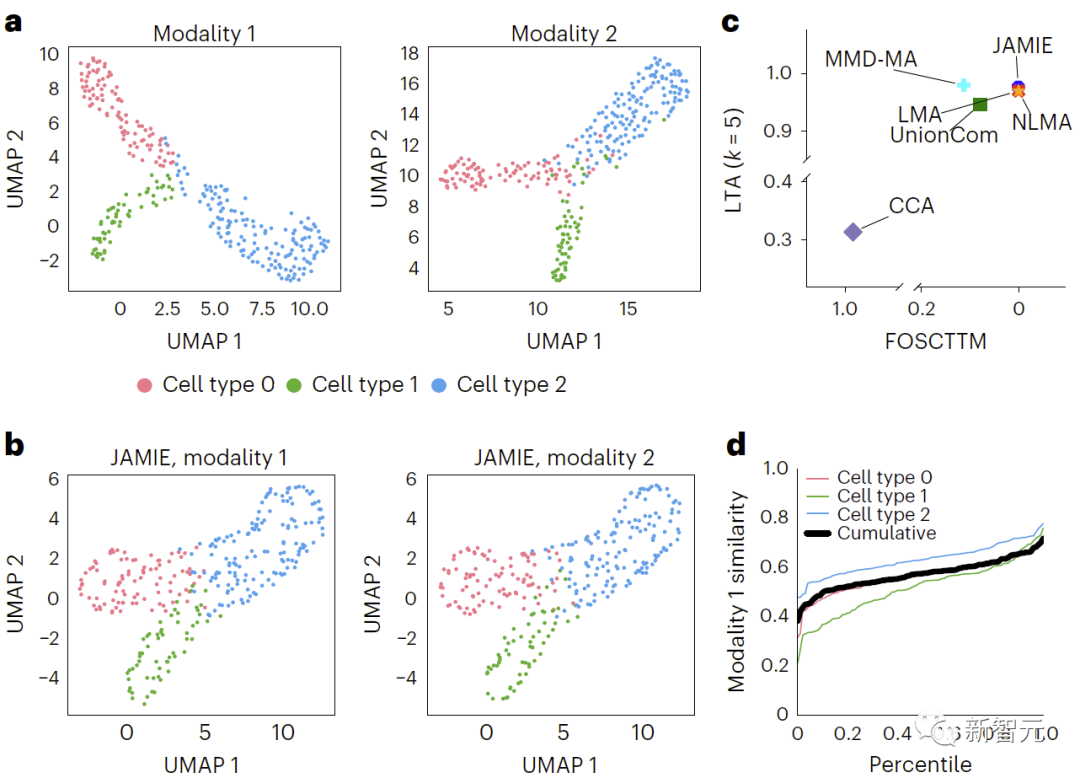
In Figure 3, by applying the UMAP algorithm to the raw spatial data and coloring according to different cell types , compared the simulated multimodal data results. b. UMAP of JAMIE latent space. c. JAMIE and existing techniques (CCA[15,16], LMA[15], MMD-MA[8], NLMA[15] and UnionCom[7]) when using all available correspondence information for cell type separation. Compare. The x-axis is the proportion of samples closer to the true mean, and the y-axis is the LTA[7,19] value. In mode 1, the cumulative distribution of the 1-JS distance is calculated to evaluate the similarity of the measured and interpolated values. Each colored line represents the similarity of a specific cell type, while the black line represents the average similarity across cell types.
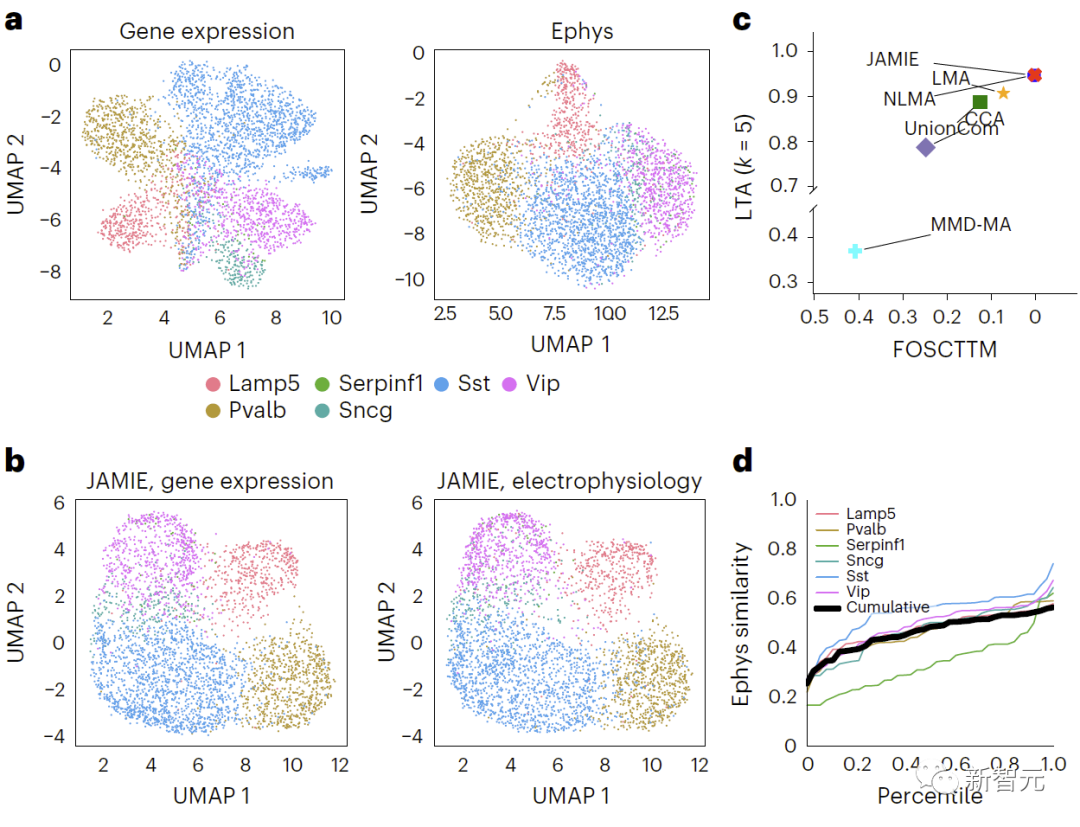
Restated: Comparing the results of gene expression and electrophysiological characteristics in mouse visual cortex, Different cell types are colored using UMAP in original space. Figure 4 shows the comparison results. b. UMAP of JAMIE latent space. c. JAMIE and existing techniques (CCA[15,16], LMA[15], MMD-MA[8], NLMA[15] and UnionCom[7]) when using all available correspondence information for cell type separation. Compare. The x-axis is the proportion of samples closer to the true mean, and the y-axis is the LTA[7,19] value. In mode 1, the cumulative distribution of the similarity between measured and interpolated values calculated from the 1-JS distance is studied. Each colored line represents the similarity of one cell type, while the black line represents the average similarity of different cell types.
In summary, JAMIE is a new deep neural network model for integrated prediction of single-cell multi-modal data.
It is suitable for complex, mixed or partially corresponding multi-modal data through a novel latent embedding aggregation method that relies on a joint variational autoencoder (VAE) structure. In addition to the above-mentioned superior performance, JAMIE also has efficient computing capabilities and low memory usage requirements. Additionally, pretrained models and learned cross-modal latent embeddings can be reused in downstream analyses.
Of course for larger data sets, training variational autoencoders (VAEs) takes a lot of time. Therefore, previous feature selection methods such as automatic PCA in JAMIE help alleviate the time requirements. Since VAE uses reconstruction loss, data preprocessing is also crucial to avoid large or repeated features from disproportionately affecting low-dimensional embedded features. For a specific cross-modal imputation, the diversity of the training data set must be carefully considered to avoid biasing the final model and negatively affecting its generalization ability. JAMIE can also potentially be extended to align data sets from different sources rather than different modalities, such as gene expression data under different conditions.
The authors of the paper are Noah Cohen Kalafut (doctoral student in the Department of Computer Science), Huang Xiang (senior researcher), and Wang Daifeng (PI) are affiliated with the University of Wisconsin-Madison Department of Biostatistics and Medical Informatics, Department of Computer Science, and Weisman Research Center. The corresponding author is Professor Wang Daifeng.
Founded in 1973, the Weisman Center has been advancing research in human development, neurodevelopmental disorders and neurodegenerative diseases for half a century.
The above is the detailed content of The UW-Chinese team's new multi-modal data analysis and generation method JAMIE greatly improves cell type and function prediction capabilities. For more information, please follow other related articles on the PHP Chinese website!
 How to bind data in dropdownlist
How to bind data in dropdownlist
 Why can't I delete the last blank page in word?
Why can't I delete the last blank page in word?
 How to share a printer between two computers
How to share a printer between two computers
 Formal digital currency trading platform
Formal digital currency trading platform
 How the temperature sensor works
How the temperature sensor works
 Solution to no sound in win7 system
Solution to no sound in win7 system
 How to solve 500 internal server error
How to solve 500 internal server error
 Mobile phone secondary card
Mobile phone secondary card








![[Web front-end] Node.js quick start](https://img.php.cn/upload/course/000/000/067/662b5d34ba7c0227.png)



