



Editor | ScienceAI
Modern healthcare systems generate large amounts of high-dimensional clinical data (HDCD), such as pulmonary function maps, photoplethysmography (PPG), electrocardiogram (ECG) recordings, CT scans, and MRI imaging , these data cannot be summarized by a single binary or continuous number.
Understanding the connection between our genome and HDCD will not only improve our understanding of the disease but will also be critical to the development of treatments for the disease.
Recently, the genomics team at Google Research has made progress in using HDCD to characterize diseases and biological characteristics.
The research team proposed an unsupervised deep learning model, Representation Learning for Low-Dimensional Embedding Gene Discovery (REGLE), to discover the association between genetic variants and HDCD.
REGLE, as a novel gene discovery method, can exploit hidden information in high-dimensional clinical data, is computationally efficient, does not require disease labels, and can integrate information from expert-defined knowledge.
Overall, REGLE contains clinically relevant information beyond what is captured by existing expert-defined signatures, allowing for improved gene discovery and disease prediction.
Related research was titled "Unsupervised representation learning on high-dimensional clinical data improves genomic discovery and prediction" and was published in "Nature Genetics" on July 8.

Paper link: https://www.nature.com/articles/s41588-024-01831-6
Study on the connection between genes and HDCD A simple approach is to perform a GWAS on each data coordinate. For example, you can study changes in the value of each pixel in a medical image. This approach is computationally expensive and has low power to detect significant associations due to high correlations between neighboring coordinates and a large multiple testing burden.
A more common approach is to focus on a small number of expert-defined features (EDF) extracted from HDCD as target features or phenotypes for GWAS. EDF can include clinically known features such as forced vital capacity (FVC) from spirometry or forced expiratory volume in 1 second (FEV1).
While these EDFs are important features discovered by experts, it is assumed that they may not fully capture the signals encoded in HDCD, so running GWAS on these signals may not exploit the full potential of HDCD.
REGLE aims to overcome these limitations using variational autoencoders (VAE) models. The method consists of three main steps:
(1) Learn the nonlinear, low-dimensional, disentangled representation (i.e., encoding or embedding) of HDCD through VAE;
(2) Conduct a GWAS independently for each encoded coordinate;
(3) Use polygenic risk scores (PRS) from coding coordinates as genetic scores for general biological functions, and then potentially combine these scores to create PRSs for specific diseases or traits (given a small number of disease labels).
Notably, REGLE also allows relevant EDFs to be selectively included in the input of the decoder in the modified VAE architecture, thus encouraging the encoder to learn only residual signals not represented by EDFs.

The researchers demonstrated the power of REGLE using two high-dimensional clinical data modalities: spirometry, which measures lung function, and spirometry, which measures cardiovascular function. PPG. Both can be collected non-invasively and relatively cheaply in clinics or on consumer wearable devices, and both modalities have well-known characteristics).
Compared with genome-wide association studies with spirometry and PPG signatures of the same dimensions, REGLE's study of learned coding recovered the majority of known genetic loci (loci) associated with lung and circulatory function, while also detecting to other sites (for example, the important site of PPG increased by 45%). If these sites are validated in further analyzes and wet lab experiments, they have the potential to become new drug targets.
A polygenic risk score (PRS) is a summary of the estimated impact of many genetic variants on a specific trait, expressed as a single number. PRSs created by genome-wide association studies on REGLE embeddings can be combined using only a few disease signatures to generate a PRS for that specific disease.
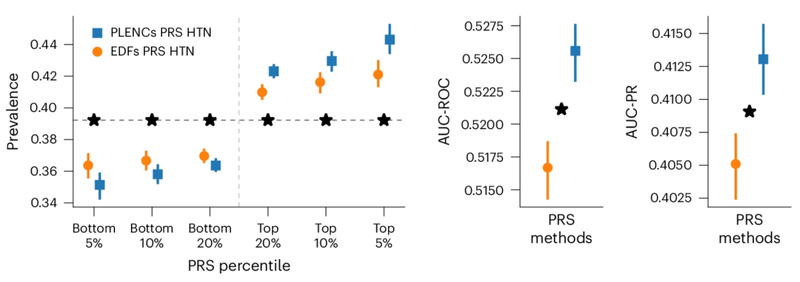
研究人员观察到,与现有方法(例如由专家定义的特征、PCA 和PRS)相比,由肺量图编码创建的肺功能PRS 改善了 COPD 和哮喘预测,并且比风险谱两端的特征PRS更有效地对风险组进行分层。哮喘和COPD 的多个独立数据集(COPDGene、eMERGE III、Indiana Biobank 和EPIC-Norfolk)中的多个指标(AUC-ROC、AUC-PR 和Pearson 相关性)在统计学上显着改善,如下所示。

类似地,从 PPG 的 REGLE 嵌入中得到的 PRS 可以改善高血压和收缩压 (SBP) 预测。在三个独立数据集(COPDGene、eMERGE III 和 EPIC-Norfolk)以及英国生物库的保留测试集中评估了由 PPG 编码和 PPG 特征生成的高血压和 SBP PRS。
观察到,在多个数据集中,使用来自 PPG 编码的 PRS 比使用来自专家定义特征的 PRS 具有一致的改进趋势,无论是高血压还是 SBP。

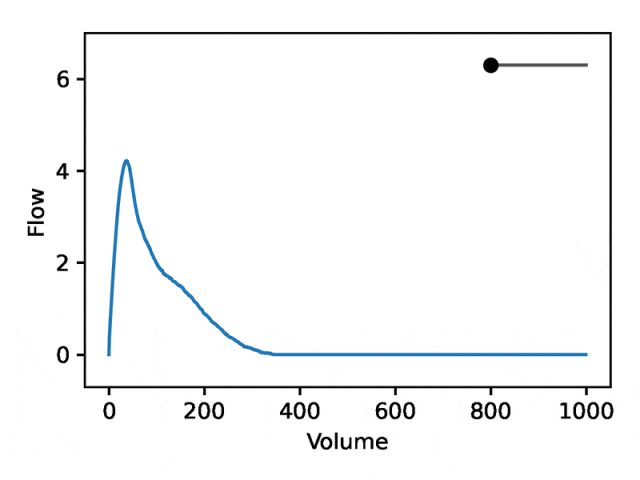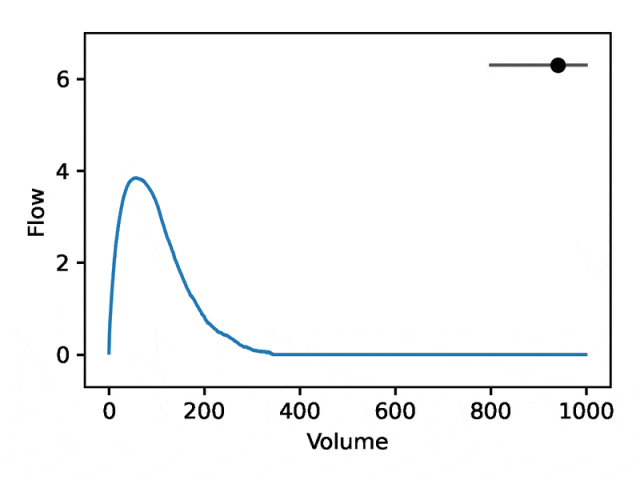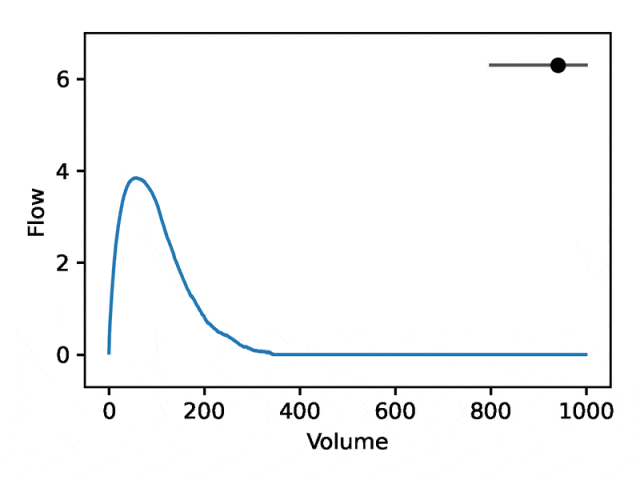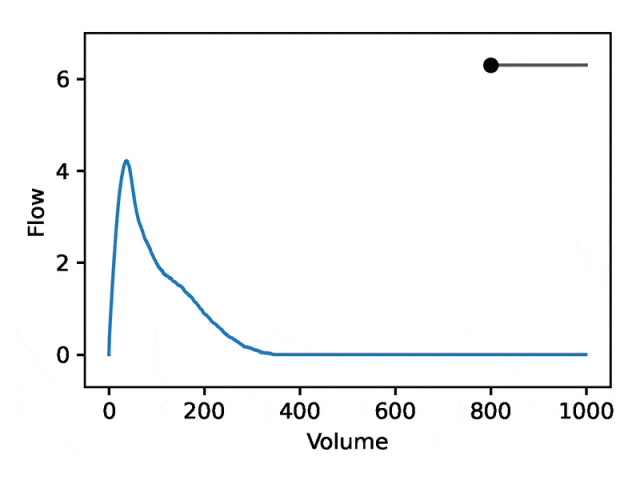
利用REGLE 的生成特性,通过固定专家定义特征的值并改变一个编码坐标而将其他编码坐标保持为零来研究编码坐标对肺量图形状的影响。然后,仅使用训练模型的解码器部分生成相应的肺量图。
典型的流量-体积肺量图由两个不同的部分组成:(1)相对较短的部分以达到峰值流量,其中流量随着体积的增加而单调增加;(2)肺量图的主要部分,其中流量单调减少。
下图显示,改变第一个坐标相当于扩大或缩小第二部分(负斜率),同时保持第一部分相对固定。事实上,曲线第二部分的凹度被肺病学家称为凹陷,这是气道阻塞的指标,标准 EDF 无法很好地表示出来。
REGLE 是一种无监督学习方法,可执行遗传分析、改进的新基因位点发现和风险预测。由于难以大规模手动发现 EDF,因此无监督学习 HDCD 表示对基因组发现很有吸引力。
REGLE 框架还通过修改传统的 VAE 架构来支持在建模中原则性地使用这些特征。在两种临床数据模式(肺量图和 PPG)中展示了 REGLE,它们可以在临床环境中进行常规测量,也可以通过智能手机或可穿戴设备被动和非侵入性地测量。
REGLE 提供了一种在没有标记数据的情况下识别遗传对器官功能影响的机制,并允许将专家特征纳入模型。它还提供了一种使用很少的标签创建疾病和特征特异性 PRS 的方法。未来,这种类似的方法将越来越多地用于进一步阐明人类特征和疾病的遗传基础。
参考内容:https://research.google/blog/harnessing-hidden-genetic-information-in-clinical-data-with-regle/
The above is the detailed content of With high efficiency and no need for labels, the Google team uses AI to mine clinical data, improve gene discovery and disease prediction, and is published in the Nature sub-journal. For more information, please follow other related articles on the PHP Chinese website!
















![[Web front-end] Node.js quick start](https://img.php.cn/upload/course/000/000/067/662b5d34ba7c0227.png)



