


编辑 | KX
此前,Google DeepMind 研究人员开发的费米子神经网络 (FermiNet) 非常适合对大量电子的量子基态进行建模。
FermiNet 最初专注于分子的基态。但是,当分子和材料受到大量能量的刺激时,例如暴露在光或高温下,电子可能会被踢入更高的能量状态——激发态。
激发态在物理学和化学等领域都很重要;然而,从第一原理出发对激发态特性进行可扩展、准确且稳健的计算仍然面临重要的理论挑战。
现在,DeepMind 研究人员开发了一种计算激发态的新方法,它比以前的方法更强大、更通用。该方法可以应用于任何类型的数学模型,包括 FermiNet 和其他神经网络。
所提出的方法对许多原子和分子实现了精确的激发态计算,远远优于现有的使用深度学习计算激发态特性的方法(尤其是在较大的系统上),并且可以应用于各种量子系统。
论文一作兼通讯作者 David Pfau 激动发文「这是深度学习首次准确解决量子物理学中一些最难的问题。希望朝着深度学习的通用量子模拟迈出新的一步。」

相关研究以「Accurate computation of quantum excited states with neural networks」为题,登上 Science !
论文链接:https://www.science.org/doi/abs/10.1126/science.adn0137
分子激发态
当分子和材料受到大量能量的刺激时,例如暴露在光或高温下,它们的电子就会被踢入一种临时的新结构,称为激发态。
分子在状态之间转换时吸收和释放的确切能量量为不同的分子和材料创建了独特的指纹。这影响了从太阳能电池板和 LED 到半导体和光催化剂等技术的性能。它们还在涉及光的生物过程中发挥着关键作用,包括光合作用和视觉。
然而,这种指纹极难建模,因为激发电子本质上是量子的,这意味着它们在分子中的位置永远不确定,只能用概率来表示。
FermiNet 可以在一系列具有各种不同定性正电子结合特性的原子和小分子中产生高度精确的、在某些情况下是最先进的基态能量。
然而 FermiNet 最初专注于分子的基态。但是,当分子和材料受到大量能量的刺激时,例如暴露在光或高温下,电子可能会被踢入更高的能量状态——激发态。
准确计算激发态的能量比计算基态能量要困难得多。即使是基态化学的黄金标准方法,如耦合簇,在激发态上也显示出数十倍的误差。虽然研究人员想将其在 FermiNet 上的工作扩展到激发态,但现有方法的效果不足以使神经网络与最先进的方法相媲美。
更强大、更通用的激发态计算新方法
DeepMind 提出了一种通过变分蒙特卡罗估计量子系统激发态的算法,该算法没有自由参数,也不需要对状态进行正交化,而是将问题转化为寻找扩展系统基态的问题。可以计算任意可观测量,包括非对角期望,例如跃迁偶极矩。
该方法特别适用于神经网络分析,通过将该方法与 FermiNet 和 Psiformer ansatz 相结合,可以准确地恢复一系列分子的激发能量和振荡器强度。

图示:从锂到氖的第一行原子的激发态能量。NES-VMC 应用于 FermiNet 的结果。(来源:论文)
研究人员将神经网络 ansätze 的灵活性与数学洞察力相结合,使其能够将寻找系统激发态的问题转换为寻找扩展系统基态的问题,然后可以使用标准 VMC 来解决。这种方法称为自然激发态 VMC (NES-VMC)。
激发态的线性独立性是通过 ansatz 的函数形式自动施加的。每个激发态的能量和其他可观测量都是通过将单态 ansätze 上的汉密尔顿期望值矩阵对角化而得到的,这些可观测量可以累积而无需额外成本。
重要なのは、この方法には調整すべき自由なパラメーターがなく、直交化を強制するペナルティ項もありません。研究者らは、FermiNet と Psiformer という 2 つの異なるニューラル ネットワーク アーキテクチャを使用して、このアプローチの精度を検証しました。
単一原子からベンゼンまで
研究者らは、単一原子からベンゼンサイズの分子までを対象としたベンチマークシステムでメソッドをテストしました。第一列原子に対する NES-VMC の精度は実験結果に非常に近いことが検証されており、一連の小分子に対しては、既存の最良の理論上のデバイス強度に匹敵する高精度のエネルギーと振動が得られます。
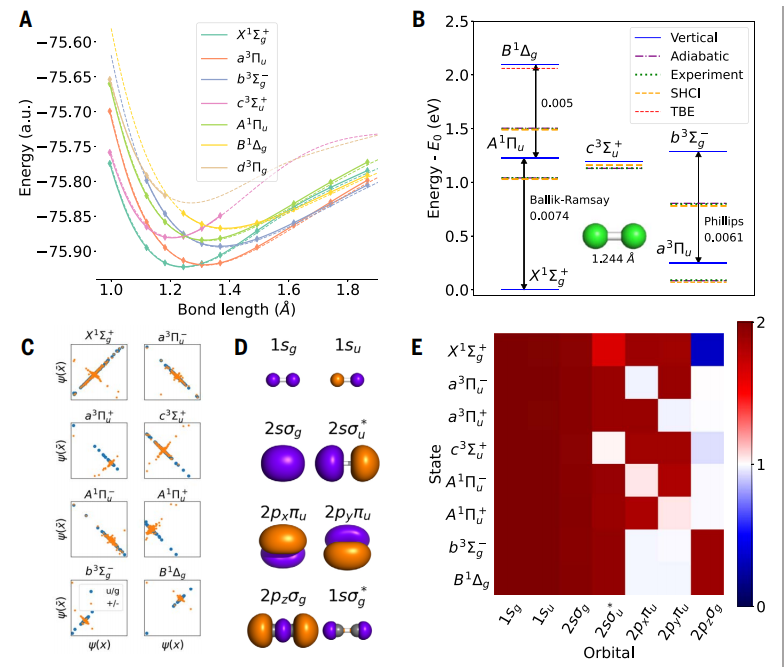
イラスト: 炭素二量体の励起状態。 (出典: 論文)
炭素二量体と呼ばれる小さく複雑な分子で 4 meV の平均絶対誤差 (MAE) を達成しました。これは、以前のゴールドスタンダード計算よりも 5 倍正確です。

イラスト: エチレンの励起状態と円錐交差。 (出典: 論文)
エチレンの場合、NES-VMC はねじれた分子の円錐交差を正確に記述し、高精度の複数参照配置相互作用 (MR-CI) の結果とよく一致します。

図: より大きな二重励起システムの励起状態。 (出典: 論文)
この研究では、複数のベンゼンクラスの分子を含む、低位二重励起を伴う 5 つの困難なシステムも検討されました。すべてのメソッドが垂直励起エネルギーに関して良好な一致を示しているシステムでは、Psiformer はブタジエンを含むすべての状態で化学的に正確であり、そのうちのいくつかの順序については数十年にわたって議論の的となっています。
数年前の最先端の計算が不正確であることが知られているテトラジンとシクロペンタジエノンの場合、NES-VMC の結果は、最近の高度な拡散モンテカルロ (DMC) および完全に活性な空間の 3 次摂動と一致しません。理論 (CASPT3) の計算は非常に近いものです。

イラスト: ベンゼンの励起状態。 (出典: 論文)
最後に、ベンゼン分子についても研究しました。NES-VMC と Psiformer ansatz を組み合わせた場合、ペナルティ法を使用したニューラル ネットワーク ansatz などの他の方法と比較して、理論上の最良推定値で優れた結果が得られました。これは、提案された方法の数学的正しさを検証するだけでなく、ニューラル ネットワークが現在の計算方法の限界で分子の励起状態を正確に表現できることも示しています。
将来の多体量子力学への応用方法
NES-VMC は、パラメーターフリーで数学的に健全な励起状態変動原理です。これをニューラル ネットワーク分析と組み合わせると、広範囲のベンチマーク問題で大幅な精度を達成できます。
量子システムの励起状態に対する正確な VMC 法は、多くの可能性を開き、ニューラル ネットワークの波動関数の適用範囲を大幅に拡大します。
この研究は分子システムとニューラルネットワークの電子励起のみを考慮しましたが、NES-VMCはあらゆる量子ハミルトニアンとあらゆるアンザッツに適用可能であり、振動電子結合、光学バンドの理解に関する科学者の理解を向上させる正確な計算研究を可能にします。ギャップ、核物理学、その他の困難な問題。
研究者らは、「NES-VMCとディープニューラルネットワークが将来、多体量子力学の最も困難な問題にどのように適用されるかを楽しみにしています
参考コンテンツ:
https:/」と述べています。 /x.com/pfau/status/1826681648597135464
https://deepmind.google/discover/blog/ferminet-quantum-physics-and-chemistry-from-first-principles/
https:// www. Imperial .ac.uk/news/255673/ai-tackles-most-difficult-challenges-quantum/
以上是AI首次解决量子物理学难题,DeepMind精确计算量子激发态,登Science的详细内容。更多信息请关注PHP中文网其他相关文章!





















