


编辑 | ScienceAI
通过运用Transformer等先进的序列模型,将单步逆合成预测问题转化为从产物SMILES表示到反应物SMILES表示的翻译任务,已经成为一种广泛采用且效果显着的策略。
然而,这种方法往往忽略了一个关键点:在反应物和产物之间,存在大量可以被直接利用的相同子结构。对这些子结构利用的不充分限制了模型预测的效率和准确性。
2024年7月,上海交通大学人工智能研究院金耀辉、许岩岩研究团队在《Journal of Cheminformatics》上发表文章《Ualign: pushing the limit of template-free retrosynthesis prediction with unsupervised SMILES alignment》。
在研究中作者提出一套单步逆合成预测流程,该流程集成了一种无监督的SMILES序列对齐技术,旨在提高化学反应预测的准确性和效率。实验结果证明了模型在预测逆合成路径方面的有效性,并表明该模型有潜力成为药物发现的有价值的工具。

Graph to Sequence的模型架构
如果将原子视作节点,化学键视为边,分子结构便能自然地转化为图结构。相较于序列模型,图神经网络更能捕捉分子内部的拓扑结构信息,从而实现更为精确的分子表征。
此外,与其他图结构相比,化学分子中的化学键携带了丰富的化学属性信息。
基于这些优势,作者提出了一种基于图注意力网络(Graph Attention Network)的变体,用以替代Transformer模型中的编码器部分,旨在为下游应用提供更为强大的分子表征能力。
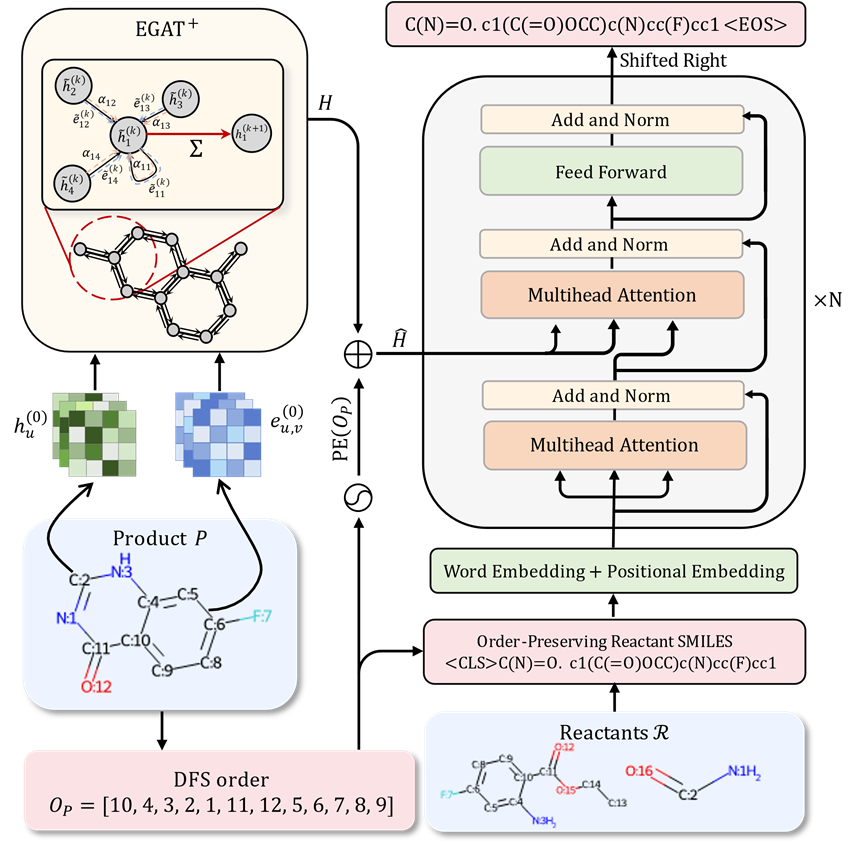
无监督的SMILES对齐机制
在单步逆合成预测中,采用序列建模的方法通常意味着必须从零开始构建反应物的结构,而不能在已有产物的基础上进行直接修改,以高效地利用反应物和产物之间相同的子结构。这种做法在一定程度上限制了生成结果的准确性。
考虑到序列建模常用的分子SMILES表征实际上是按照深度优先搜索的顺序排列分子中的原子和化学键,如果能向模型提供每个产物原子在反应物SMILES表征中出现的位置信息,将有助于模型识别并复用在反应过程中未发生改变的子结构。这将显着降低模型预测反应物的难度,并提高预测的准确性。
从序列建模的角度来看,常用的分子SMILES表征实质上是依据深度优先搜索(DFS)的顺序对分子中的原子和化学键进行排列。若能向模型提供产物中每个原子在反应物SMILES表征中的位置信息,将极大地促进模型对未发生改变的子结构的识别和复用,从而显着降低预测反应物的难度,提高预测的准确性。
然而,直接提供这种对应信息可能会在模型训练过程中引入信息泄露的风险。为避免这一问题,研究者提出了一种创新的策略,旨在不泄露标签信息的前提下,优化模型对反应物分子结构的理解和预测能力。
考虑到SMILES序列表征源自于分子图上的深度优先搜索,并且反应物与产物间的大部分子结构具有高度一致性,对于给定的任意产物的DFS顺序,必然存在一个与之对应的反应物分子图上的DFS顺序,使得反应物和产物上的对应原子出现的顺序几乎相同。
基于此策略,研究者不仅将产物分子结构纳入模型输入,还引入了反应物分子的DFS顺序作为输入的一部分。
此外,研究者根据上述策略生成了与给定反应物DFS顺序高度一致的产物分子DFS顺序,并利用这一顺序生成反应物的SMILES表征,作为模型训练的目标。
这种设计使得反应物和产物之间的相似子结构在模型的输入和输出中以几乎相同的顺序排列,从而简化了模型学习反应物和产物相同结构对应关系的过程,并有助于识别反应过程中发生改变的基团。
即便在从零开始构建反应物结构的情况下,这种方法也能有效地复用产物结构信息,显着提高预测的准确率。
尤为重要的是,由于产物的DFS顺序仅基于其分子结构信息,不依赖于任何关于反应物的信息作为标注,这种方法有效避免了模型训练过程中的标签泄露问题。
同时,这种无监督的SMILES对齐方式在训练过程中无需引入额外的监督信号,从而避免了复杂的数据标注和多任务学习中的优化难题,为分子逆合成预测领域提供了一种新颖且高效的研究途径。
实验结果展示
在本项研究中,作者对多个分子逆合成预测数据集进行了系统性评估,涵盖了广泛使用的USPTO-50K数据集,以及数据量更庞大的USPTO- MIT和USPTO-FULL。
评估模型性能时,采用了top-k准确率作为主要的评价指标。在USPTO-50K数据集上,作者不仅考察了模型生成的SMILES序列的合法性,还通过大规模预训练的正向反应预测模型,对模型输出的合成方案进行了实际可行性的回环验证。
表1:USPTO-50K逆合成预测的Top-k准确率
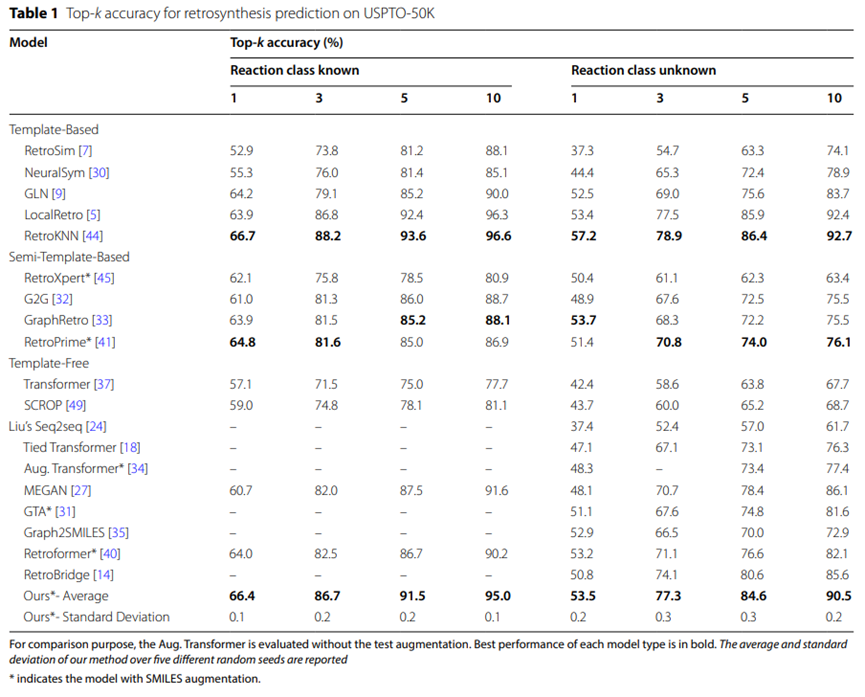
USPTO-50K数据集的实验结果汇总于表1,显示在未指定具体反应类型的条件下,UAlign模型在USPTO -50K数据集上的top-5准确率高达84.6%,显着优于其他无模板的基线模型。
表2:USPTO-MIT逆合成预测的Top-k准确率
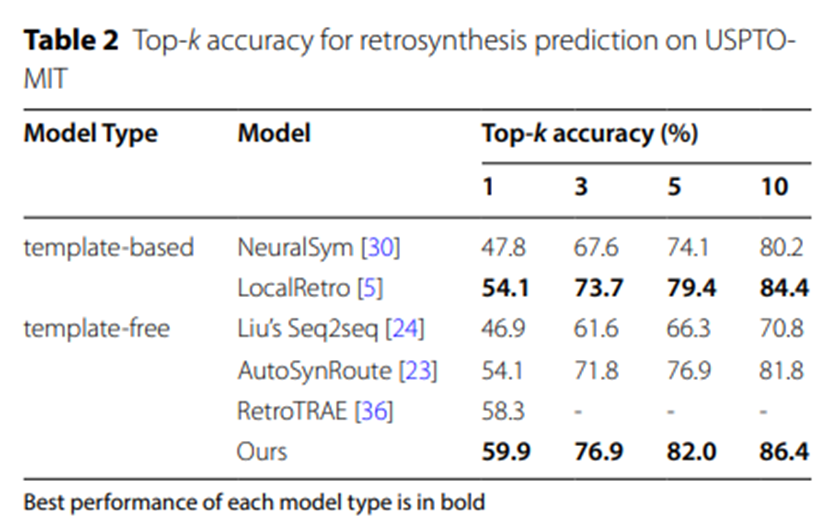
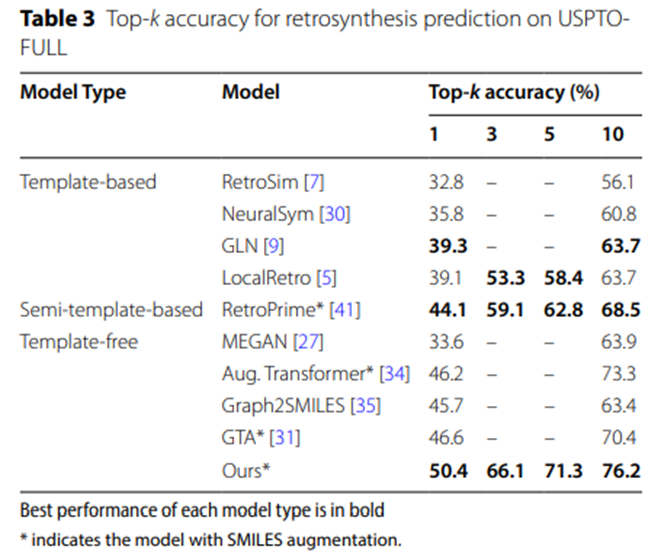
表2和表3的实验数据进一步证实,在更大规模的数据集USPTO-MIT和USPTO-FULL上,UAlign模型以显着的优势超越了其他各类基线模型。
表3:USPTO-FULL上逆合成预测的Top-k准确率
此外,表4的实验结果表明,与其他基于SMILES的逆合成预测模型相比,UAlign模型生成的反应物SMILES序列具有更高的合法性。
表4:在USPTO-50K上对反应类别未知的逆合成预测的Top-k SMILES 有效性
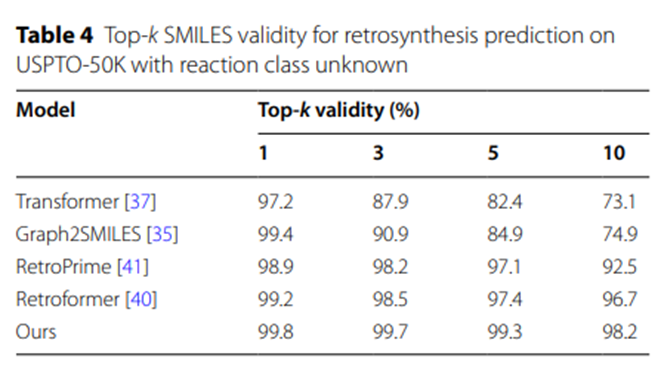
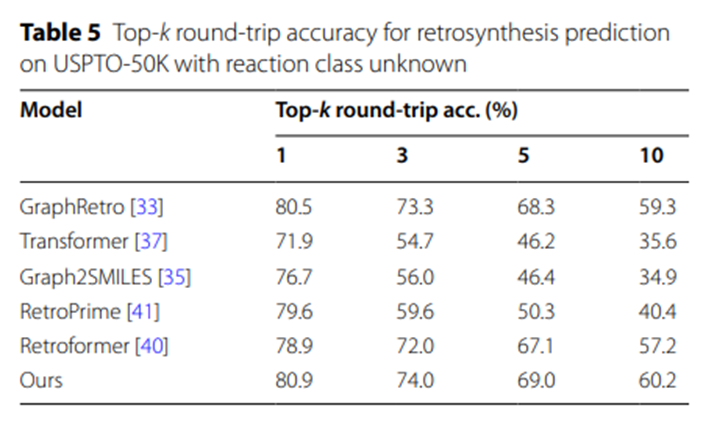
表5的实验数据进一步凸显了UAlign模型在生成合理且可行的合成方案方面的优势。原因在于,UAlign所提出的合成方案中,有较高比例的方案能够通过正向反应预测模型的验证,即这些方案在经过相应的化学反应后,能够有效地转化为给定的目标产物。
表5:USPTO-50K上反应类别未知的逆合成预测Top-k往返准确率
这些实验结果不仅验证了UAlign模型在分子逆合成预测任务中的高效性和准确性,也突显了其在处理大规模数据集时的卓越性能和在生成高质量合成方案方面的显着优势。
为验证UAlign模型在实际生产中的应用潜力,作者选取了近两年内由美国食品药品监督管理局(FDA)批准上市的新药作为合成目标,通过模型的多次迭代调用,成功获取了合成路线。模型对这两种药物合成路线的预测结果与文献中记录的路径高度一致。
此外,对于第三种药物,模型所预测的合成路线也得到了化学领域专家的可行性认可。这些合成路径不仅涵盖了多种反应类型,还包含了诸如环状化合物的合成以及涉及多个反应中心的单步逆合成预测等复杂情况。
上述实验结果充分证明了UAlign模型不仅能够应对多样化的反应类型,而且在实际生产中具有较高的应用价值。这表明UAlign模型在分子逆合成预测领域具有强大的实用性和灵活性,能够为药物合成提供有效的解决方案。

未来展望
UAlign模型凭借其卓越的性能和灵活性,完全有能力作为构建多步逆合成系统的基石。它能够与各种搜索算法以及多目标优化技术相结合,形成一套高效、智能的逆合成路径规划系统。
此外,作者也在积极探索将UAlign算法与先进的硬件设备相融合,以打造自动化的无人实验室来推进药物发现和合成过程的自动化,为化学研究和药物开发领域带来革命性的变革。
以上是化学逆合成SOTA!上海交大团队提出SMILES对齐技术实现高效逆合成预测的详细内容。更多信息请关注PHP中文网其他相关文章!





















