



La post-fonctionnalisation est une méthode économique pour optimiser les propriétés des candidats médicaments. Cependant, la complexité chimique des molécules médicamenteuses rend souvent la fonctionnalisation à un stade avancé difficile.
Pour résoudre ce problème, des chercheurs de l'Université de Munich, de l'ETH Zurich et de Roche Bâle ont collaboré pour développer une plateforme de fonctionnalisation à un stade avancé. La plateforme est basée sur la géométrie. Technologie d'apprentissage profond et de criblage de réactions à haut débit
Étant donné que la borylation est l'une des étapes clés de la fonctionnalisation, nous avons utilisé des modèles informatiques pour prédire le rendement dans différentes conditions de réaction avec une plage d'erreur absolue moyenne de 4 à 5 %. Le modèle a pu classer de nouvelles réactions pour des substrats connus et inconnus avec une précision de 92 % et 67 % respectivement. Nous avons pu capturer avec précision la régiosélectivité du produit principal, avec un score F de 67 % pour le classificateur. Lorsqu'elle a été appliquée à 23 molécules médicamenteuses commerciales différentes, nous avons découvert avec succès de nombreuses opportunités de diversification structurelle. la revue Nature Chemistry du 23 mars

La nouveauté des structures lorsqu'on vise à établir des relations structure-activité en chimie médicinale et la complexité rendent la synthèse de structures cibles chimiques difficile. Les modèles de relation structure-activité peuvent guider les composés principaux et diriger les plans d’optimisation des composés pour améliorer l’activité pharmacologique et les propriétés physicochimiques des candidats médicaments. Pour l'exploration des relations structure-activité, une intégration efficace est cruciale, ce qui constitue le goulot d'étranglement du cycle conception-réalisation-test-analyse
Il existe de nombreuses méthodes alternatives pour activer et modifier les liaisons C-H pour la fonctionnalisation tardive des échafaudages organiques ( LSF), allant des éléments de base moléculaires aux molécules médicamenteuses avancées. De nombreux systèmes catalytiques proposent des approches directionnelles et non directionnelles, ainsi qu'un accès chimique et sélectif sur site à des analogues modifiés.
Parmi les nombreuses méthodes LSF, la méthode de borylation C-H est considérée comme la méthode la plus couramment utilisée pour une diversification rapide des composés. Les composés organoborés peuvent être convertis en une variété de groupes fonctionnels comme moyen fiable pour les réactions ultérieures de couplage de liaisons C-C, permettant ainsi des recherches approfondies sur les relations structure-activité. Cependant, à l'heure actuelle, il n'existe que quelques applications du LSF dans le rapport sur la découverte de médicaments. La plupart de ces rapports se concentrent sur un seul type de réaction LSF. Le LSF direct de plusieurs types de liaisons C – H avec différentes forces de liaison, propriétés électroniques et environnements de groupes stériques et fonctionnels pose des défis. De plus, mener des projets LSF demande souvent beaucoup de temps et de ressources, ce qui n'est pas cohérent avec les calendriers serrés et les actifs limités de nombreux projets de chimie médicinale
Le graphique montre un aperçu de la recherche sur la diversification de la boronisation. (Source des données : papier)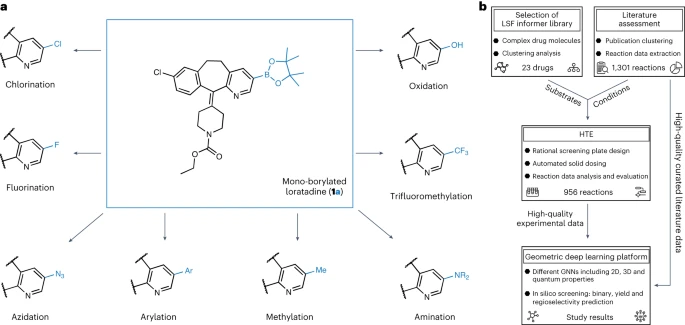
L'expérimentation à haut débit (HTE) est une méthode établie d'optimisation des réactions qui permet une miniaturisation semi-automatique et un criblage de petits lots, permettant une optimisation rapide des réactions. , Exécutez de manière reproductible plusieurs transformations en parallèle en utilisant un petit nombre de blocs de construction et de consommables précieux. Combiné à la documentation FAIR (Findability, Accessibility, Interoperability, Reusability) qui génère des ensembles de données de haute qualité sur les réponses réussies et échouées, HTE permet une analyse avancée des données et un apprentissage automatique pour débloquer LSF pour la découverte de médicaments Foundation. Le réseau neuronal graphique (GNN) a été largement utilisé dans l'extraction de caractéristiques moléculaires et la prédiction d'attributs. Parmi les diverses méthodes d'apprentissage automatique développées pour la planification des réactions chimiques, les GNN ont été appliqués avec succès à la planification rétrosynthétique, à la prédiction de la régiosélectivité et à la prédiction des produits de réaction. De plus, des transformateurs et des méthodes basées sur les empreintes digitales ont également été développés pour résoudre des problèmes similaires.
La recherche a montré qu'en apprenant la géométrie de l'état de transition, le résultat d'une réaction concurrente peut être prédit avec précision. Les prédictions de la régiosélectivité des réactions provoquées par des effets électroniques peuvent être améliorées à l'aide de la théorie fonctionnelle de la densité (DFT) et de la caractérisation graphique des charges partielles atomiques. En combinant l'apprentissage automatique graphique et les expériences à haut débit (HTE), les conditions des réactions d'activation C-H des substrats organiques peuvent être optimisées. Certaines recherches se sont concentrées sur l'utilisation de modèles d'apprentissage profond des états de transition qui ont la capacité de prédire les résultats des réactions, y compris l'énantiosélectivité dans certains cas.
Cependant, ces méthodes sont limitées à de petites structures moléculaires et à des ensembles de données relativement petits, ce qui rend difficile la recherche. appliquer de tels modèles à des molécules de type médicament plus structurellement complexes. Sur la base de recherches documentaires, la régiosélectivité des réactions de borylation catalysées par l'iridium peut être prédite grâce à un modèle hybride d'apprentissage automatique amélioré par des informations chimiques quantiques sur les états de transition. Cependant, l'impact des effets stériques et électroniques n'a pas encore été exploré sur les performances des modèles de réactions d'activation C-H et leurs applications régiosélectives dans des molécules comportant plusieurs systèmes de cycles aromatiques
Criblage automatisé de la boronisation LSF avec apprentissage profond géométrique
Des chercheurs de l'Université de Munich, de l'ETH Zurich et de Roche Pharmaceuticals Basel présentent une méthode automatisée de criblage de la boronisation LSF appliquée à l'apprentissage profond géométrique pour identifier les opportunités de diversification des hits et des leads à un stade avancé. L’apprentissage informatique profond a été utilisé pour prédire le résultat de la réaction, le rendement et la régiosélectivité des molécules médicamenteuses complexes LSF.
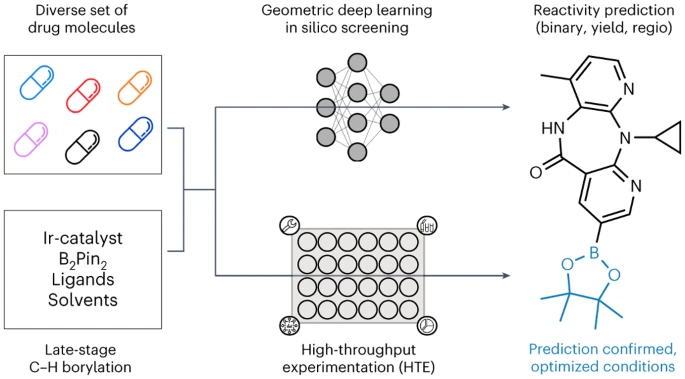
David Nippa, auteur principal et doctorant dans le groupe de recherche de l'École de chimie et de pharmacie de LMU et Roche, a déclaré qu'avec cette approche, le nombre d'expériences en laboratoire pourrait être considérablement réduit, augmentant ainsi l'efficacité de synthèse chimique et durabilité
Pour les premières étapes de cette recherche, nous avons effectué une analyse approfondie de la littérature publiée afin de sélectionner des conditions de réaction de criblage à haut débit appropriées et des substrats pertinents pour les propriétés des composés principaux de la découverte de médicaments à un stade avancé. Nous avons déterminé les conditions de réaction sur la base d'un ensemble de données de 38 publications organisées manuellement. La sélection du substrat LSF était basée sur les résultats d'une analyse groupée de 1 174 médicaments approuvés, aboutissant à 23 molécules médicamenteuses structurellement différentes. Cette approche permet aux chercheurs d'utiliser des exemples pertinents de conditions de réaction et de substrats dans une approche de « bibliothèque d'informations », plutôt que de s'appuyer uniquement sur des substrats idéaux et des fragments d'applicabilité limitée pour optimiser la synthèse du composé principal.
Dans la seconde Dans cette étape, les chercheurs utilisent des semi -des expériences automatisées à haut débit (HTE) pour générer des données (ensembles de données expérimentales). Les données de réaction des molécules médicamenteuses sélectionnées et des conditions de réaction fournissent des données de haute qualité pour l'apprentissage automatique des résultats de réaction ultérieurs.
Enfin, nous avons formé différents modèles de réseaux neuronaux graphiques (GNN), en utilisant une charge partielle bidimensionnelle, tridimensionnelle et atomique. renforcement Utilisez des graphiques moléculaires pour prédire les résultats des réactions binaires, les rendements des réactions et les régiosélectivités. "Il est intéressant de noter que les prévisions se sont améliorées lorsque nous avons pris en compte les informations tridimensionnelles sur le matériau de départ plutôt que sa formule chimique bidimensionnelle", a déclaré Kenneth Atz, doctorant à l'ETH Zurich.
Cette approche a été utilisée avec succès pour identifier les positions. au sein des ingrédients actifs existants où des groupes réactifs supplémentaires peuvent être introduits. Cela aidera les chercheurs à développer plus rapidement de nouvelles variantes plus efficaces d'ingrédients actifs pharmaceutiques connus
Veuillez cliquer sur le lien suivant pour consulter le contenu de l'article : https://www.nature.com/articles/s41557-023- 01360-5
Rapports associés : https://techxplore.com/news/2023-11-Artificial Intelligence ouvre la voie aux médicaments.html
Ce qui précède est le contenu détaillé de. pour plus d'informations, suivez d'autres articles connexes sur le site Web de PHP en chinois!
 Type d'utilisation en JavaScript
Type d'utilisation en JavaScript
 Une liste complète des commandes alter dans Mysql
Une liste complète des commandes alter dans Mysql
 Comment télécharger le pilote de la souris Razer
Comment télécharger le pilote de la souris Razer
 Que dois-je faire si mon iPad ne peut pas être chargé ?
Que dois-je faire si mon iPad ne peut pas être chargé ?
 Comment configurer Douyin pour empêcher tout le monde de voir l'œuvre
Comment configurer Douyin pour empêcher tout le monde de voir l'œuvre
 tutoriel pascal
tutoriel pascal
 commande de recherche de fichier Linux
commande de recherche de fichier Linux
 Les fichiers de programme peuvent-ils être supprimés ?
Les fichiers de programme peuvent-ils être supprimés ?




















![[Web front-end] Démarrage rapide de Node.js](https://img.php.cn/upload/course/000/000/067/662b5d34ba7c0227.png)



