


In den letzten Jahren konnten wir mit der rasanten Entwicklung der Einzelzelltechnologie verschiedene Eigenschaften einzelner Zellen messen, um multimodale Einzelzelldaten zu erhalten (wie scRNA-seq, scATAC-seq, Patch-seq). ).
Diese Daten helfen uns, ein tieferes Verständnis zellulärer Funktionen und molekularer Mechanismen zu erlangen. Beispielsweise haben Forscher kürzlich Methoden des maschinellen Lernens eingesetzt, um die Beziehung zwischen multimodalen Daten einzelner Zellen zu analysieren und die biologischen Mechanismen zu verstehen, die an Zelltypen und Krankheiten beteiligt sind.
Allerdings ist die Erfassung multimodaler Einzelzellendaten oft kostspielig und es kommt häufig zu Modalverlusten. Bestehende Methoden des maschinellen Lernens erfordern in der Regel vollständig abgestimmte multimodale Daten zum Füllen und Einbetten von Daten und sind nicht für Situationen geeignet, in denen Modalitäten fehlen.
Um dieses Problem zu lösen, entwickelte Wang Daifengs Labor an der University of Wisconsin-Madison eine Open-Source-Methode für maschinelles Lernen, die auf gemeinsamen Variations-Autoencodern basiert – Joint Variational Autoencoders for Multimodal Imputation and Embedding (JAMIE).
JAMIE kann für die integrierte Analyse multimodaler Einzelzellendaten verwendet werden, wie z. B. Datenausrichtung, Einbettung und Ergänzung fehlender Daten, um Zelltypen und -funktionen besser vorherzusagen.
Diese Arbeit wurde kürzlich in Nature Machine Intelligence veröffentlicht.

Papieradresse: https://www.nature.com/articles/s42256-023-00663-z
Projektadresse: https://github.com/daifengwanglab /JAMIE: Einführung in die JAMIE-Methode
JAMIE kombiniert die wiederverwendbare und flexible Latentraumgenerierung von Autoencodern mit der automatischen Korrespondenzschätzung von Ausrichtungsmethoden und ermöglicht so die Verarbeitung multimodaler Daten mit unvollständiger Korrespondenz.
Abbildung 1. Übersicht über die JAMIE-Methode
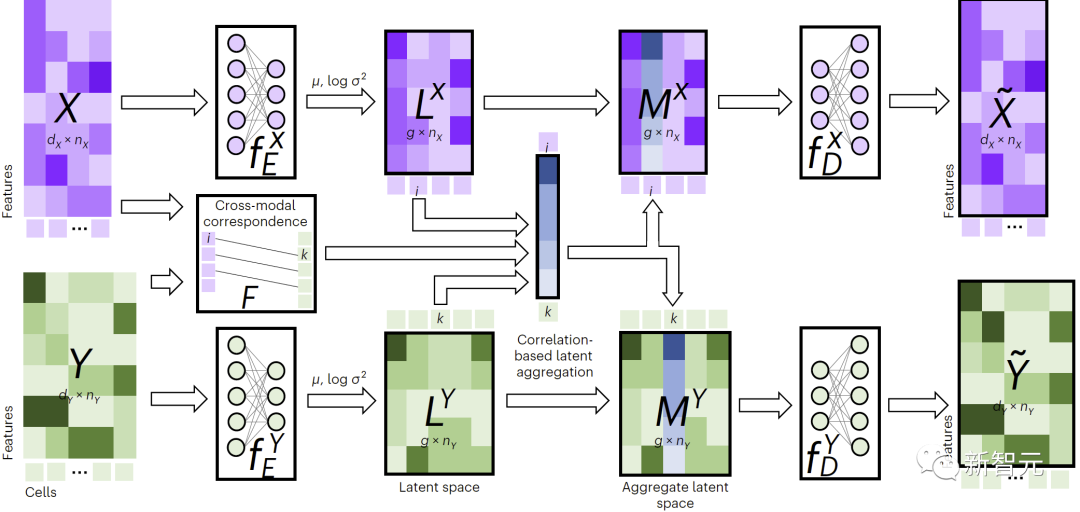
Im Einzelnen kann JAMIE in die folgenden zwei Schritte unterteilt werden: Vorverarbeitung der Eingabedaten. Nehmen wir als Beispiel den bimodalen Modus und gehen davon aus, dass die den Modi entsprechenden Datenmatrizen bzw. sind. Beachten Sie, dass hier die Merkmalsabmessungen und die Summe unterschiedlich sein können und auch die Anzahl der Stichproben unterschiedlich sein kann. Durch die Vorverarbeitung wird jede Zeile jeder Matrix so normalisiert, dass sie einen Mittelwert von 0 und eine Varianz von 1 aufweist. Wenn entsprechende Daten vorhanden sind, kann der Benutzer eine modale Korrelationsmatrix bereitstellen, um die Leistung zu verbessern. Dies bedeutet, dass die Stichprobe im Modal vollständig der Stichprobe im Modal entspricht, dass keine bekannte Entsprechung vorliegt und dass eine teilweise Übereinstimmung vorliegt Korrespondenz.
Verwenden Sie den gemeinsamen Variations-Autoencoder, um den latenten Ähnlichkeitsraum jeder Modalität zu lernen: und , wobei (Standard, vom Benutzer anpassbar) die Dimension des latenten Raums ist. Während des Trainingsprozesses minimiert JAMIE die folgende Verlustfunktion:
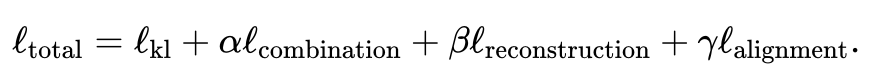
Für spezifische Ausdrücke der einzelnen Punkte sehen Sie sich bitte den Originaltext des Papiers an. Die Gewichtungen des zweiten, dritten und vierten Elements im Verhältnis zum ersten Element können vom Benutzer angepasst werden. JAMIE bietet auch Standardgewichtungen, die für häufige Situationen geeignet sind.
Die folgende Tabelle zeigt den Vergleich des Modells und Anwendungsbereichs von JAMIE mit den aktuellen State-of-the-Art-Methoden. JAMIE vereint die Funktionen mehrerer verschiedener Integrations- und Interpolationsmethoden in einer einzigen Architektur und ermöglicht so die Interpolation fehlender Modalitäten, die Kompatibilität von Nicht-Omics-Daten und die Möglichkeit, multimodale Daten mit nur teilweiser Übereinstimmung zu verarbeiten.
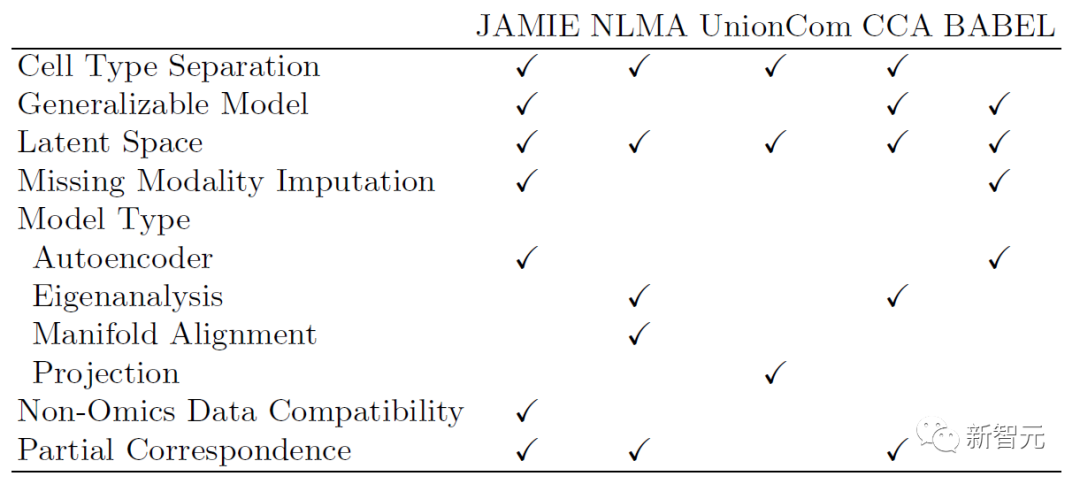
Tabelle 1. Vergleich verschiedener multimodaler Integrations- und fehlender modaler Füllmethoden. Durch eine einzige Architektur integriert JAMIE Funktionen aus mehreren verschiedenen Integrations- und Interpolationsmethoden. NLMA: Nonlinear Manifold Alignment [15], UnionCom [7], CCA: Canonical Correlation Analysis [15, 16], BABEL [5].
Integration und Phänotypvorhersage multimodaler Daten
Die Integration multimodaler Daten kann die Klassifizierungsleistung verbessern, das phänotypische Wissen und das Verständnis komplexer biologischer Mechanismen verbessern.
Angesichts zweier Datensätze und entsprechender Beziehungen kann JAMIE latente Raumdaten basierend auf dem trainierten Encoder generieren und basierend darauf Clustering oder Klassifizierung durchführen.
Clustering basierend auf latenten Raumdaten hat mehrere Vorteile, wie zum Beispiel die Einbeziehung beider Modalitäten in die Feature-Generierung. JAMIE kann dann Probenkorrespondenzen vorhersagen, beispielsweise die Vorhersage des Zelltyps.
Bei teilweise beschrifteten Datensätzen sollten Zellen im selben Cluster ähnliche Typen haben.
JAMIE trennt die Eigenschaften verschiedener Datentypen im Prozess der Generierung latenter Raumdaten, sodass komplexe Clustering- oder Klassifizierungsalgorithmen normalerweise nicht erforderlich sind, um bessere Ergebnisse zu erzielen.
Für hochdimensionale Daten verwendet JAMIE UMAP [32] zur Visualisierung der Zelltyp-Clusterbildung.
Kreuzmodale Datenimputation
Viele aktuelle kreuzmodale Imputationsmethoden können nicht nachweisen, dass sie die zugrunde liegenden biologischen Mechanismen für Imputationszwecke gelernt haben.
Im Vergleich zu Feedforward-Netzwerken oder linearen Regressionsmethoden kann JAMIE die zugrunde liegenden biologischen Mechanismen besser erlernen, um fehlende Daten auf der Grundlage strengerer mathematischer Grundlagen vorherzusagen.
Abbildung 2 zeigt JAMIES Prozess zur modalübergreifenden Datenbefüllung. JAMIE trainiert zunächst die Kodierungs- und Dekodierungsmodelle anhand der Trainingsdaten.
Für neue Daten verwendet JAMIE zunächst den aus den Daten erlernten Encoder, um sie in den latenten Raum zu projizieren, um sie zu erhalten, erhält sie dann durch Aggregation latenter Raummerkmale und dekodiert sie schließlich über den entsprechenden Decoder in fehlende Musterdaten.
JAMIE nutzt latenten Raum, um die Korrespondenz zwischen Zellen vorherzusagen, was dabei helfen kann, die Beziehung zwischen Datenmerkmalen und Phänotypen zu verstehen.
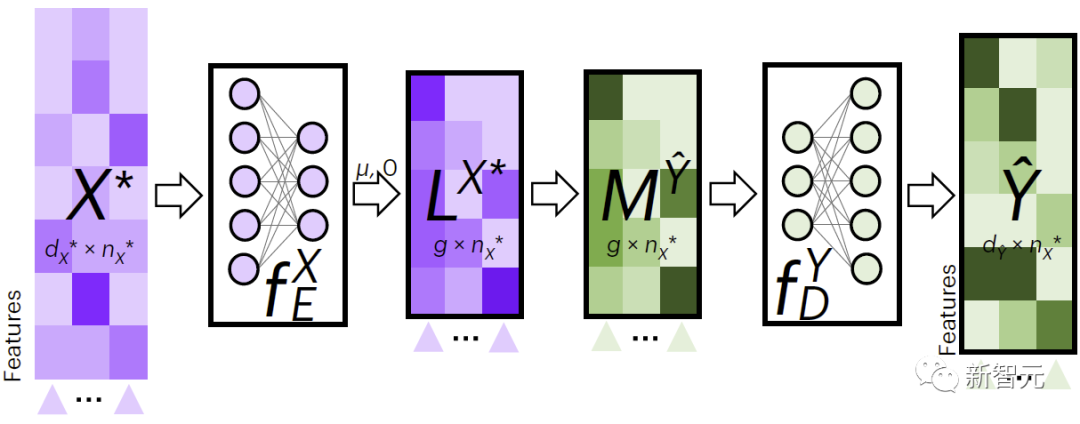
Abbildung 2. JAMIE-Cross-Modal-Interpolation
Erklärung der latenten Raummerkmale und Füllmerkmale
Um das zu erklären Modell übernimmt JAMIE SHAP (SHapley Additive Erläuterungen)[18].
SHAP bewertet die Bedeutung einzelner Eingabemerkmale durch Stichprobenmodulation der einzelnen vom Modell generierten Vorhersagen. Dies kann für eine Vielzahl interessanter Anwendungen genutzt werden.
Wenn die Zielvariable leicht nach Phänotyp getrennt werden kann, kann SHAP relevante Merkmale für weitere Untersuchungen identifizieren. Wenn wir außerdem eine Imputation durchführen, kann SHAP die vom Modell gelernten modalübergreifenden Verbindungen aufdecken.
Lernen Sie anhand eines Modells und eines Beispiels den SHAP-Wert, sodass sich der Hintergrundmerkmalsvektor befindet.
Wenn , dann ist die Summe der SHAP-Werte und der Hintergrundausgabe gleich, wobei jeder proportional zur Auswirkung auf die Modellausgabe ist.
Eine weitere nützliche Technik besteht darin, eine Schlüsselmetrik für die Klassifizierung (z. B. LTA [7, 19]) oder die Imputation (z. B. Korrespondenz zwischen unterstellten Merkmalen und gemessenen Merkmalen) auszuwählen und sie einzeln im Modell zu verwenden. Die Metrik ist ausgewertet, indem jedes Merkmal entfernt (durch Hintergrundwerte ersetzt) wird.
Wenn sich dann die Schlüsselmetrik verschlechtert, deutet dies darauf hin, dass die entfernten Features für die Ergebnisse des Modells wichtiger sind.
JAMIE verwendete zur Verifizierung vier häufig verwendete multimodale Einzelzellen-Datensätze.
(1) Simulierte multimodale Daten (300 Proben, 3 Zelltypen), generiert durch Gaußsche Verteilungsstichprobe verzweigter Mannigfaltigkeiten aus MMD-MA;
(2) aus dem visuellen Kortex der Maus (Patch-seq-Genexpression und elektrophysiologische Daten). Charakterisierungsdaten einzelner neuronaler Zellen im motorischen Kortex der Maus (1.208 Proben, 9 Zelltypen) und im motorischen Kortex der Maus (1.208 Proben, 9 Zelltypen); Zugänglichkeitsdaten für 8.981 Proben im sich entwickelnden menschlichen Gehirn (21 Schwangerschaftswochen, die 7 Hauptzelltypen in der menschlichen Großhirnrinde abdecken); die COLO-320DM-Colon-Adenokarzinom-Zelllinie.
Die Auswertung ergab, dass JAMIE deutlich besser ist als andere Methoden (Vergleich der Ergebnisse der Verzweigungsmannigfaltigkeitssimulationsdaten von MMD-MA in Abbildung 3 und Vergleich der Ergebnisse der visuellen Cortex-Daten der Maus in Abbildung 4) und der multimodalen Methode Vorrang einräumt Füllen wichtiger Funktionen und Bereitstellung potenziell neuer mechanistischer Erkenntnisse bei zellulärer Auflösung.
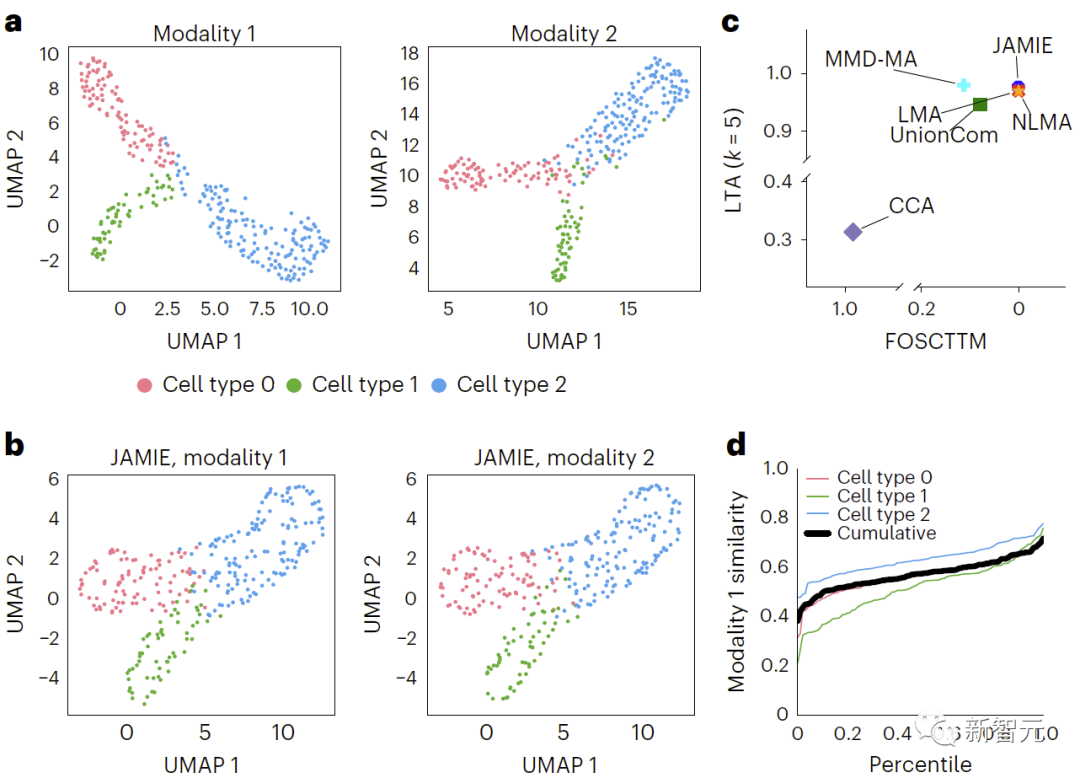
In Abbildung 3 werden die simulierten multimodalen Datenergebnisse verglichen, indem der UMAP-Algorithmus auf die räumlichen Rohdaten angewendet und entsprechend verschiedenen Zelltypen eingefärbt wird. b. UMAP des JAMIE-Latentraums. c. JAMIE und bestehende Techniken (CCA[15,16], LMA[15], MMD-MA[8], NLMA[15] und UnionCom[7]) bei Verwendung aller verfügbaren Korrespondenzinformationen für die Zelltyptrennung. Die x-Achse ist der Anteil der Stichproben, die näher am wahren Mittelwert liegen, und die y-Achse ist der LTA[7,19]-Wert. Im Modus 1 wird die kumulative Verteilung des 1-JS-Abstands berechnet, um die Ähnlichkeit der gemessenen und interpolierten Werte zu bewerten. Jede farbige Linie stellt die Ähnlichkeit eines bestimmten Zelltyps dar, während die schwarze Linie die durchschnittliche Ähnlichkeit zwischen den Zelltypen darstellt.
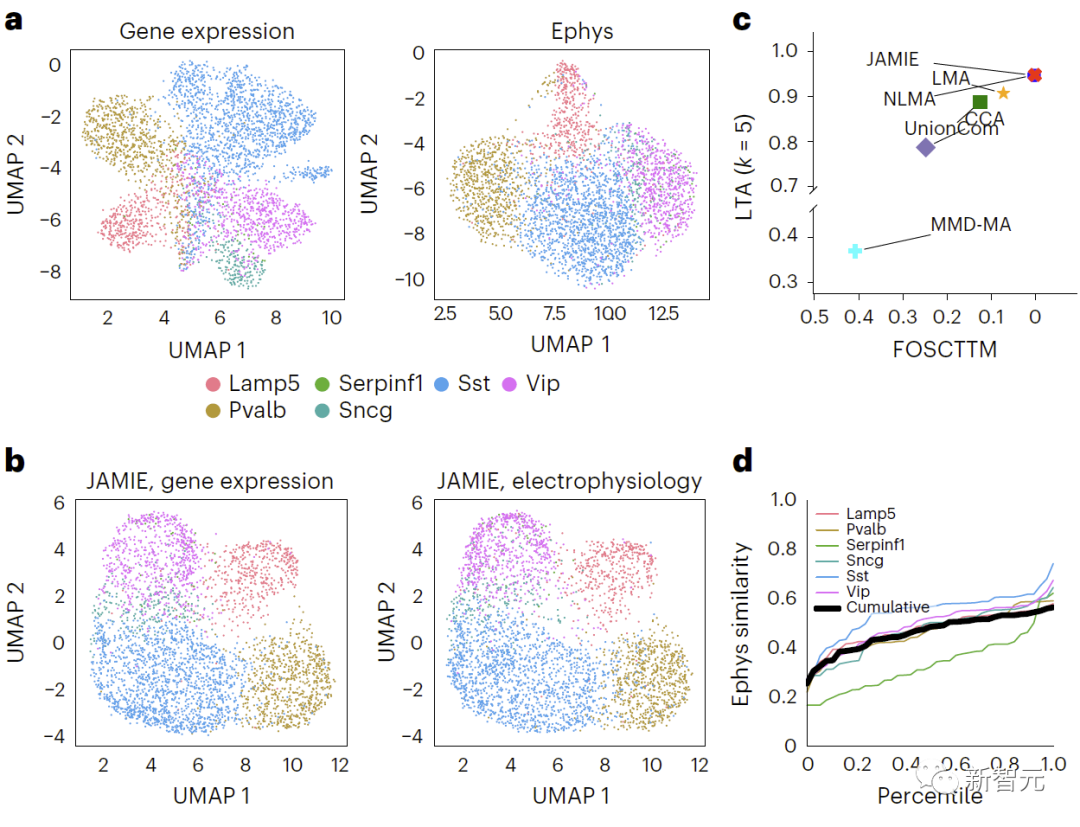
Neu formuliert: Der Vergleich der Genexpression und der elektrophysiologischen Signatur führt zu Ergebnissen im visuellen Kortex der Maus, wobei UMAP im ursprünglichen Raum verwendet wird und verschiedene Zelltypen gefärbt werden. Abbildung 4 zeigt die Vergleichsergebnisse. b. UMAP des JAMIE-Latentraums. c. JAMIE und bestehende Techniken (CCA[15,16], LMA[15], MMD-MA[8], NLMA[15] und UnionCom[7]) bei Verwendung aller verfügbaren Korrespondenzinformationen für die Zelltyptrennung. Die x-Achse ist der Anteil der Stichproben, die näher am wahren Mittelwert liegen, und die y-Achse ist der LTA[7,19]-Wert. Im Modus 1 wird die kumulative Verteilung der Ähnlichkeit zwischen gemessenen und interpolierten Werten untersucht, die aus der 1-JS-Distanz berechnet werden. Jede farbige Linie stellt die Ähnlichkeit eines Zelltyps dar, während die schwarze Linie die durchschnittliche Ähnlichkeit verschiedener Zelltypen darstellt.
Zusammenfassend ist JAMIE ein neues tiefes neuronales Netzwerkmodell für die integrierte Vorhersage multimodaler Einzelzellendaten. Es eignet sich für komplexe, gemischte oder teilweise entsprechende multimodale Daten und wird durch eine neuartige latente Einbettungsaggregationsmethode implementiert, die auf einer Joint Variational Autoencoder (VAE)-Struktur basiert. Neben der oben genannten überlegenen Leistung verfügt JAMIE auch über effiziente Rechenfunktionen und einen geringen Speicherbedarf. Darüber hinaus können vorab trainierte Modelle und erlernte modalübergreifende latente Einbettungen in nachgelagerten Analysen wiederverwendet werden. Natürlich nimmt das Training von Variational Autoencodern (VAEs) bei größeren Datensätzen viel Zeit in Anspruch. Daher tragen frühere Methoden zur Funktionsauswahl wie die automatische PCA in JAMIE dazu bei, den Zeitaufwand zu verringern. Da VAE Rekonstruktionsverluste verwendet, ist die Datenvorverarbeitung auch von entscheidender Bedeutung, um zu verhindern, dass große oder wiederholte Features unverhältnismäßig große oder wiederholte Features beeinträchtigen und niedrigdimensionale eingebettete Features beeinträchtigen. Für eine spezifische modalübergreifende Imputation muss die Vielfalt des Trainingsdatensatzes sorgfältig berücksichtigt werden, um eine Verzerrung des endgültigen Modells und eine negative Auswirkung auf seine Generalisierungsfähigkeit zu vermeiden. JAMIE kann möglicherweise auch erweitert werden, um Datensätze aus unterschiedlichen Quellen anstelle unterschiedlicher Modalitäten, wie etwa Genexpressionsdaten unter unterschiedlichen Bedingungen, abzugleichen. Die Autoren des Papiers sind Noah Cohen Kalafut (Doktorand im Fachbereich Informatik), Huang Xiang (leitender Forscher) und Wang Daifeng (PI). Abteilung für Biostatistik und Medizinische Informatik, Abteilung für Informatik, University of Wisconsin-Madison und Weisman Research Center. Der korrespondierende Autor ist Professor Wang Daifeng. Das 1973 gegründete Weisman Center treibt seit einem halben Jahrhundert die Forschung in den Bereichen menschliche Entwicklung, neurologische Entwicklungsstörungen und neurodegenerative Erkrankungen voran. Einführung in die Autoren
Das obige ist der detaillierte Inhalt vonDie neue multimodale Datenanalyse- und Generierungsmethode JAMIE des UW-Chinese-Teams verbessert die Vorhersagefähigkeiten für Zelltyp und -funktion erheblich. Für weitere Informationen folgen Sie bitte anderen verwandten Artikeln auf der PHP chinesischen Website!
 So binden Sie Daten in einer Dropdown-Liste
So binden Sie Daten in einer Dropdown-Liste
 Warum kann ich die letzte leere Seite in Word nicht löschen?
Warum kann ich die letzte leere Seite in Word nicht löschen?
 So teilen Sie einen Drucker zwischen zwei Computern
So teilen Sie einen Drucker zwischen zwei Computern
 Formelle Handelsplattform für digitale Währungen
Formelle Handelsplattform für digitale Währungen
 So funktioniert der Temperatursensor
So funktioniert der Temperatursensor
 Lösung für keinen Ton im Win7-System
Lösung für keinen Ton im Win7-System
 So beheben Sie 500 interne Serverfehler
So beheben Sie 500 interne Serverfehler
 Mobiltelefon-Zweitkarte
Mobiltelefon-Zweitkarte








![[Web-Frontend] Node.js-Schnellstart](https://img.php.cn/upload/course/000/000/067/662b5d34ba7c0227.png)



