後期功能化是一種經濟的方法,用於優化候選藥物的特性。然而,藥物分子的化學複雜性經常使得後期功能化變得具有挑戰性
為了解決這個問題,德國慕尼黑大學、蘇黎世聯邦理工學院和巴塞爾羅氏製藥的研究人員合作開發了一個後期功能化平台,該平台基於幾何深度學習和高通量反應篩選技術
鑑於硼基化是功能化的關鍵步驟之一,我們使用計算模型預測了不同反應條件下的產率,平均絕對誤差範圍為4-5%。此模型能夠對已知和未知底物進行新反應的分類,分類的準確度分別為92%和67%。我們能夠準確捕捉主要產物的區域選擇性,分類器的F分數為67%。我們在應用於23種不同的商業藥物分子時,成功發現了許多結構多樣化的機會
該研究題為“利用幾何深度學習實現高通量實驗來促進後期藥物多樣化”,於2023年11月23日在《自然化學》雜誌上發表

#LSF計畫在藥物化學研究中扮演重要的角色
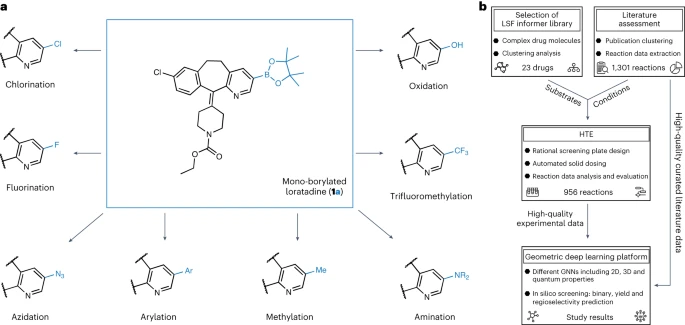
# ####在旨在建立藥物化學中的結構-活性關係時,結構的新穎性和複雜性使得合成化學目標結構具有挑戰性。構效關係模型可指導先導化合物和先導化合物最佳化方案,以提高候選藥物的藥理活性和理化性質。對於結構-活性關係的探索,高效整合至關重要,這是設計-製造-測試-分析週期的瓶頸######有許多可選擇的方法可以激活和修飾C-H鍵,以實現有機支架的後期功能化(LSF),範圍從分子構件到高級藥物分子。許多催化系統提供了定向和非定向的方法,以及對修飾類似物的化學和位點選擇性訪問######在眾多的LSF方法中,C-H硼化方法被認為是最常用的快速化合物多樣化方法。有機硼化合物可以轉化為多種官能基,作為後續的C-C鍵偶聯反應的可靠手段,從而實現廣泛的結構-活性關係研究#######然而,目前來看,在藥物發現中,LSF的應用僅有一些報道。這些通報中大多數都集中在單一的LSF反應類型上。對於具有不同鍵強度、電子特性以及空間和官能基環境的多種類型的C-H鍵直接進行LSF提出了挑戰。此外,開展LSF專案通常需要耗費大量的時間和資源,這與許多藥物化學專案的緊迫時間表和有限資產並不相符##############圖表展示了硼化多樣化研究的概述。 (資料來源:論文)#########人工智慧支援的LSF(Language Support Feature)#########高通量實驗(HTE) 是既定的反應優化方法,可實現半自動小型化小批量篩選,從而快速、可重複地使用少量珍貴的建造模組和耗材並行執行多個轉換。結合可產生有關成功和失敗反應的高品質資料集的FAIR(可查找性、可訪問性、互通性、可重用性)文檔,HTE 透過實現高級資料分析和機器學習,為解鎖LSF 進行藥物發現奠定了基礎。 ######圖神經網路(GNN)在分子特徵提取和屬性預測方面有著廣泛的應用。在為化學反應規劃開發的各種機器學習方法中,GNN 已成功應用於逆合成規劃、區域選擇性預測和反應產物預測。此外,還開發了 transformer 和基於指紋的方法來解決類似的問題######有研究顯示,透過學習過渡態的幾何結構,可以準確地預測競爭反應的結果。利用密度泛函理論(DFT)和原子部分電荷的圖形特徵化,可以改進對電子效應驅動的反應的區域選擇性的預測。結合圖機器學習和高通量實驗(HTE),可以優化有機底物 C-H 活化反應的條件。一些研究著重於使用過渡態的深度學習模型,這些模型具有預測反應結果的能力,包括在某些情況下的對映選擇性######然而,這些方法僅限於小分子結構和相對較小的數據集,使得將此類模型應用於結構更複雜的藥物樣分子具有挑戰性。根據文獻研究,透過過渡態的量子化學資訊增強的混合機器學習模型,可以預測銥催化的硼化反應的區域選擇性。然而,對於C-H活化反應模型的性能,以及在具有多個芳香環系統的分子中的區域選擇性應用,空間效應和電子效應的影響尚未被探索###
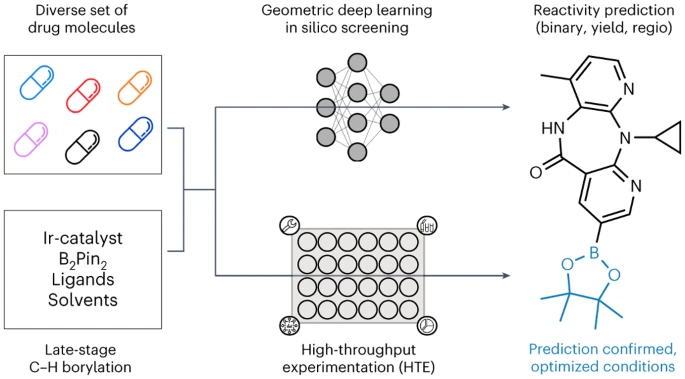
几何深度学习的自动LSF硼化筛选
慕尼黑大学、苏黎世联邦理工学院和巴塞尔罗氏制药的研究人员介绍了一种应用于几何深度学习的自动LSF硼化筛选方法,用于识别后期命中和先导多样化机会。采用计算深度学习来预测复杂药物分子 LSF 的反应结果、产量和区域选择性。
慕尼黑大学化学与药学学院和罗氏公司的研究小组主要作者、博士生David Nippa表示,通过这种方法,实验室实验的数量可能会显著减少,从而提高化学合成的效率和可持续性
对于该研究的首要步骤,我们对已经发表的文献进行了彻底的分析,以便选择适当的高通量筛选反应条件和与药物发现后期先导化合物性质相关的底物。我们根据手动整理的38篇文献数据集来确定反应条件
LSF底物的选择是基于对1,174种已批准药物的聚类分析结果,得出了23种结构不同的药物分子。这种方法使得研究人员能够在“信息库”方法中使用反应条件和底物的相关示例,而不是仅仅依赖于适用性有限的理想底物和片段来优化先导化合物的合成方法
在第二步中,研究人员使用半自动化高通量实验(HTE)生成数据(实验数据集)。所选药物分子和反应条件的反应数据为后续反应结果的机器学习提供了高质量的数据
最后,我们训练了不同的图神经网络(GNN)模型,使用二维、三维以及原子部分电荷加强的分子图来预测二元反应结果、反应产率以及区域选择性。苏黎世联邦理工学院的博士生 Kenneth Atz 表示:“有趣的是,当我们考虑起始材料的三维信息而不仅仅是其二维化学式时,预测结果得到了改善。”
这种方法已经成功地用于确定现有活性成分中可以引入额外活性基团的位置。这对于研究人员更快地开发已知药物活性成分的新的、更有效的变体具有帮助
请点击以下链接查看论文内容:https://www.nature.com/articles/s41557-023-01360-5
相关报道:https://techxplore.com/news/2023-11-人工智能为药物铺平道路.html
以上是使用幾何深度學習方法預測合成藥物分子的最佳方案,為新藥發現鋪路的詳細內容。更多資訊請關注PHP中文網其他相關文章!