


Rewritten content is: Ziluo
Large-scale simulations of complex electron interactions remain one of the greatest challenges in atomic modeling. While classical force fields often fail to describe couplings between electronic states and ion rearrangements, more accurate ab initio molecular dynamics suffer from computational complexity that prevents long and large-scale simulations, which are relevant for research techniques The phenomenon is crucial
Recently, researchers from the University of California, Berkeley, and Lawrence Berkeley National Laboratory proposed a machine learning interatomic potential (MLIP) model based on graph neural networks: the crystalline Hamiltonian graph Neural network (Crystal Hamiltonian Graph Neural Network, CHGNet) can model a universal potential energy surface.
The study highlights the importance of charge information for capturing appropriate chemical reactions and provides insights into ion systems with additional electronic degrees of freedom unobservable with previous MLIPs.
The research was titled "CHGNet as a pretrained universal neural network potential for charge-informed atomistic modeling" and was published in "Nature Machine Intelligence" on September 14, 2023.

Large-scale simulations, such as molecular dynamics (MD), are important tools for computational exploration of solid-state materials. However, accurately modeling electronic interactions and their subtle effects in molecular dynamics simulations remains a huge challenge. Empirical methods such as classical force fields are often not accurate enough to capture complex electronic interactions
Ab initio molecular dynamics (AIMD) combined with density functional theory (DFT) can be used to explicitly calculate density functional Approximating the electronic structure within, producing high-fidelity results with quantum mechanical precision. Long-term, large-scale spin-polarized AIMD simulations, critical for studying ion migration, phase transitions, and chemical reactions, are challenging and computationally expensive.
MLIPs such as ænet and DeepMD offer promising solutions for bridging the gap between expensive electronic structure methods and efficient classical interatomic potentials. However, incorporating the important effects of valence on chemical bonding remains a challenge in MLIP.
Charge can be represented in a variety of ways, from simple oxidation state labels to continuous wave functions derived from quantum mechanics. The challenge of incorporating charge information into MLIP comes from many factors, such as ambiguity of representation, complexity of interpretation, scarcity of labels, etc.
The content that needs to be rewritten is: CHGNet architecture
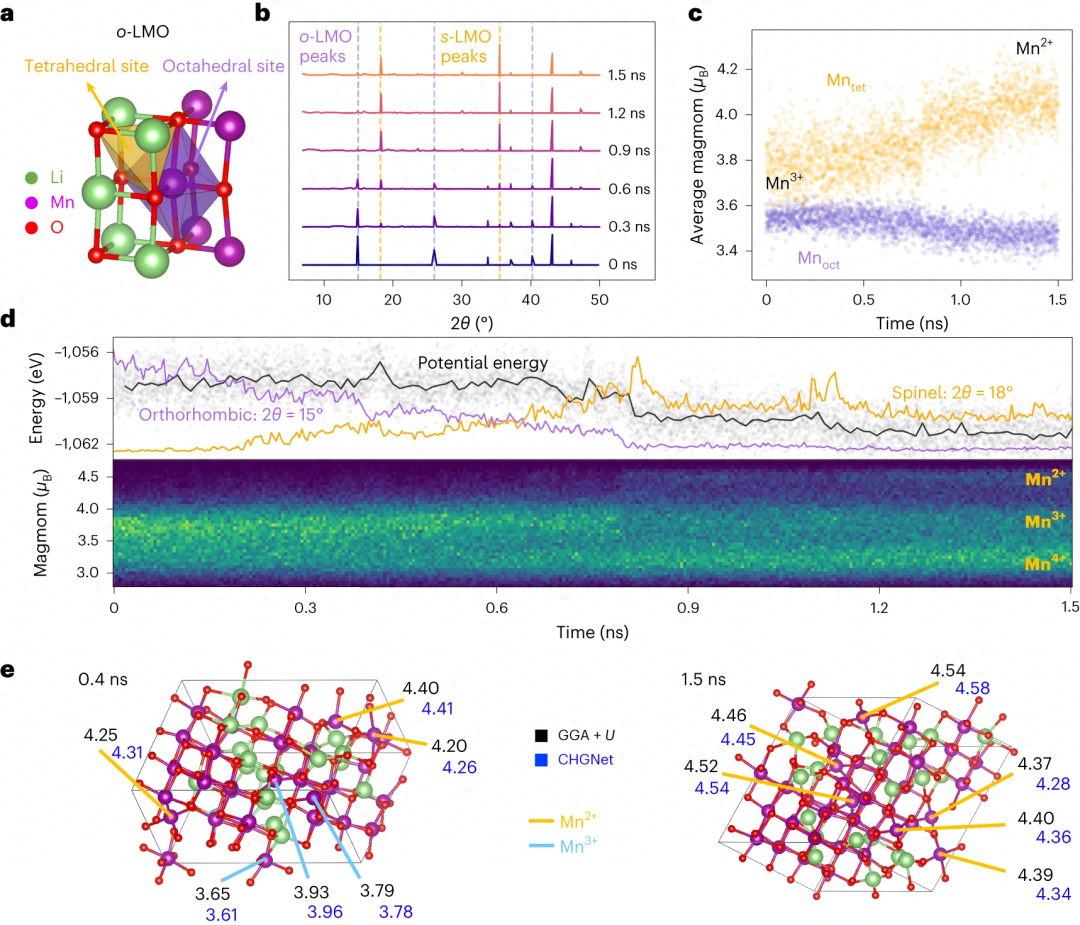
CHGNet is pre-trained on energy, force, stress and magnetic moment from the Materials Project Trajectory Dataset (MPtrj). The data set contains more than 10 years of density functional theory calculations on 1.5 million inorganic structures. By explicitly including magnetic moments, CHGNet is able to learn and accurately represent the orbital occupation of electrons, thereby enhancing its ability to describe atomic and electronic degrees of freedom
#MPtrj The distribution of elements in the data set is shown below Shown

# Rewritten content: Illustration: The distribution of elements in the MPtrj data set. (Source: Paper)
Here, the researchers define charge as an atomic property (atomic charge) that can be inferred by the inclusion of magnetic moments (magmoms). Research shows that by explicitly incorporating site-specific magmoms as charge state constraints into CHGNet, latent space regularization can be enhanced and electronic interactions can be accurately captured
CHGNet is based on GNN, where Figure Vol. Accumulated layers are used to propagate atomic information through a set of nodes {vi} connected by edges {eij}. Translation, rotation and alignment invariance are preserved in GNN. CHGNet takes as input a crystal structure with unknown atomic charges and outputs the corresponding energies, forces, stresses, and magmoms. Charge-decorated structures can be inferred from field magmoms and atomic orbital theory.

The rewritten content is as follows: Illustration: CHGNet model architecture. (Source: paper)
In CHGNet, periodic crystal structures are converted into atomic graphs  by searching for neighboring atoms vj within
by searching for neighboring atoms vj within  of each atom vi in the original unit.
of each atom vi in the original unit.
Different from other GNNs, in which the atomic features updated after t convolutional layers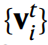 are directly used to predict energy, CHGNet regularizes the node features of t−1 convolutional layers
are directly used to predict energy, CHGNet regularizes the node features of t−1 convolutional layers
 to include information about the magma. Regularized features
to include information about the magma. Regularized features carry rich information about the local ion environment and charge distribution. Therefore, the atomic signature used to predict energy, force, and stress is the charge constrained by information about its charge state. Therefore, CHGNet can provide charge state information using only nuclear positions and atomic identities as inputs, allowing the study of charge distribution in atomic modeling.
carry rich information about the local ion environment and charge distribution. Therefore, the atomic signature used to predict energy, force, and stress is the charge constrained by information about its charge state. Therefore, CHGNet can provide charge state information using only nuclear positions and atomic identities as inputs, allowing the study of charge distribution in atomic modeling. 




Second, although magmom is a good heuristic for atomic charges calculated from spin polarization in ionic systems, it is realized that atomic charge inference for non-magnetic ions can be ambiguous, so Additional domain knowledge is required. Therefore, for ions without magmom, the atom-centered magmom cannot accurately reflect their atomic charge. CHGNet will infer the charge from the environment, similar to other MLIP functions.
can be further improved by combining other charge representation methods Enhanced models such as electron positioning functions, electric polarization and atomic orbital based partitioning. These methods can be used for atomic feature engineering in latent space
CHGNet can implement charge-based atomic simulations and is suitable for large-scale computational simulations to study heterovalent systems, thus expanding the scope of computational chemistry, physics, biology and research opportunities on charge transfer coupling phenomena in materials science
Please click the following link to view the paper: https://www.nature.com/articles/s42256-023-00716-3
The above is the detailed content of Charge-based atomic simulation implementation using pre-trained general purpose neural network CHGNet. For more information, please follow other related articles on the PHP Chinese website!
















![[Web front-end] Node.js quick start](https://img.php.cn/upload/course/000/000/067/662b5d34ba7c0227.png)



