



Proteins are involved in many biological functions such as cell composition, muscle contraction, digestion of food, and identification of viruses.
In order to design better proteins (including antibodies), scientists often repeatedly mutate amino acids (arranging the units that make up proteins in a certain order) at different positions until the protein obtains the required function.
But there are more amino acid sequences than there are grains of sand in the world, so finding the best protein, and thus the best potential drug, is often difficult. When faced with this challenge, scientists often spend millions of dollars and test on miniaturized, simplified versions of biological systems.
“This requires a lot of guesswork and verification.” Brian L. Hie, assistant professor of chemical engineering at Stanford University and innovation fellow at the Arc Institute, said, “The goal of many intelligent algorithms is to take the guesswork out of it.”
Stanford University scientists have developed a new method based on machine learning that can predict molecular changes that lead to better antibody drugs faster and more accurately. Combining the 3D structure of the protein backbone with a large language model based on the amino acid sequence, the researchers were able to find rare and desirable mutations in minutes.
The study was titled "Unsupervised evolution of protein and antibody complexes with a structure-informed language model" and was published in "Science" on July 4, 2024.

Large language models trained solely on sequence information can learn high-level principles of protein design. However, in addition to sequence, the three-dimensional structure of proteins determines their specific function, activity, and evolvability.
For antibody engineering problems, researchers at Stanford University applied structurally informed protein language models to predict high-fitness sequences constrained by known antibody or antibody-antigen complex structures.
Research shows that a universal protein language model augmented with protein structural backbone coordinates can guide the evolution of different proteins without the need to model individual functional tasks.
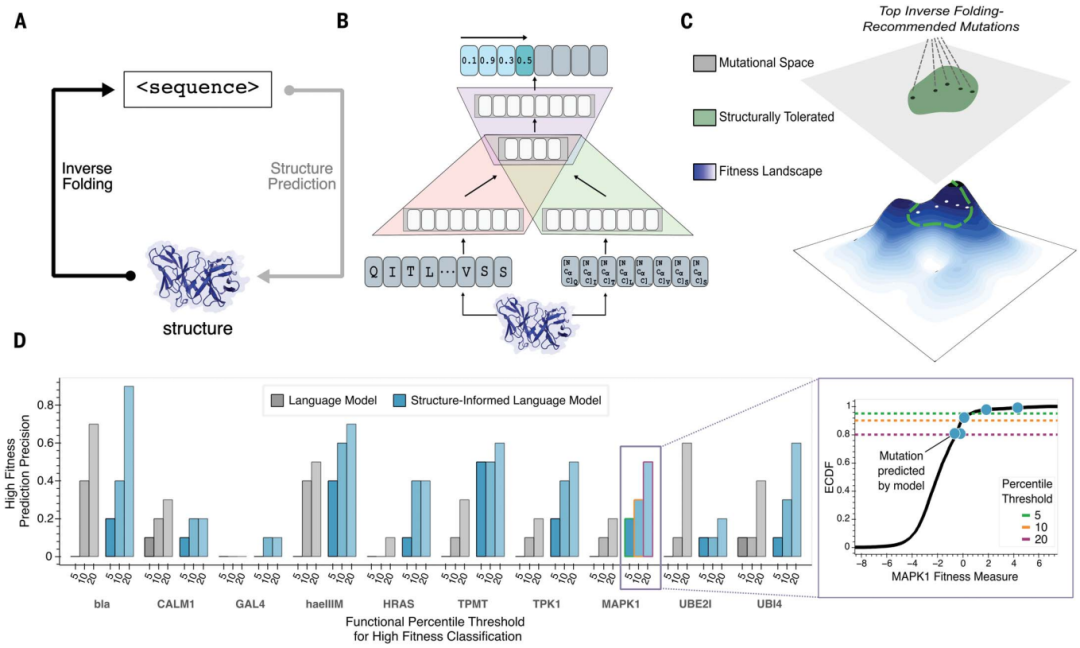
Structure-guided paradigm:
Wide application:
Protein complex design:
Human Antibody Evolution:
Replace large amounts of data:
Directed evolution:

With this method, the team screened about 30 candidates for two therapeutic clinical antibodies for the treatment of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection. Variants. At the same time, the researchers achieved a 25-fold increase in neutralization and a 37-fold increase in affinity against BQ.1.1 and XBB.1.5 antibody-escape virus variants, respectively.
In conclusion,This tool will help quickly respond to new or developing diseases. It also lowers the barriers to making more effective drugs. Stronger drugs mean lower doses are needed, meaning more patients can benefit from a given dose.
論文連結:https://www.science.org/doi/10.1126/science.adk8946
相關報告:https://phys.org/news/2024-07-ai-approach-optimizes-approach-optimizes-approach-optimizes-approach-optimizes-approach-optimizes- antibody-drugs.html
The above is the detailed content of Login to Science, drug affinity increased 37 times, AI performs unsupervised optimization of protein and antibody complexes. For more information, please follow other related articles on the PHP Chinese website!




























![[Web front-end] Node.js quick start](https://img.php.cn/upload/course/000/000/067/662b5d34ba7c0227.png)



